Category Archives: Metal
A metal (from Greek μέταλλον métallon, “mine, quarry, metal”[1][2]) is a material (an element, compound, or alloy) that is typically hard, opaque, shiny, and has good electrical and thermal conductivity. Metals are generally malleable — that is, they can be hammered or pressed permanently out of shape without breaking or cracking — as well as fusible (able to be fused or melted) and ductile (able to be drawn out into a thin wire).[3] About 91 of the 118 elements in the periodic table are metals (some elements appear in both metallic and non-metallic forms).
The meaning of “metal” differs for various communities. For example, astronomers use the blanket term “metal” for convenience to collectively describe all elements other than hydrogen and helium (the main components of stars, which in turn form most of the visible matter in the universe). Thus, in astronomy and physical cosmology, the metallicity of an object is the proportion of its matter made up of chemical elements other than hydrogen and helium.[4] In addition, many elements and compounds that are not normally classified as metals become metallic under high pressures; these are formed as metallic allotrope of non-metals.
Lanthanide
From Wikipedia, the free encyclopedia
| Lanthanides in the periodic table | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The lanthanide /ˈlænθənaɪd/ or lanthanoid /ˈlænθənɔɪd/ series of chemical elements[1] comprises the fifteen metallic chemical elementswith atomic numbers 57 through 71, from lanthanum through lutetium.[2][3][4] These fifteen lanthanide elements, along with the chemically similar elements scandium and yttrium, are often collectively known as the rare earth elements.
The informal chemical symbol Ln is used in general discussions of lanthanide chemistry to refer to any lanthanide. All but one of the lanthanides are f-block elements, corresponding to the filling of the 4f electron shell; lutetium, a d-block element, is also generally considered to be a lanthanide due to its chemical similarities with the other fourteen. All lanthanide elements form trivalent cations, Ln3+, whose chemistry is largely determined by the ionic radius, which decreases steadily from lanthanum to lutetium.
They are termed as lanthanides because the lighter elements in the series are chemically similar to lanthanum. Strictly speaking, both lanthanum and lutetium have been labeled as group 3 elements, because they both have a single valence electron in the d shell. However, both elements are often included in any general discussion of the chemistry of the lanthanide elements.
In presentations of the periodic table, the lanthanides and the actinides are customarily shown as two additional rows below the main body of the table,[2] with placeholders or else a selected single element of each series (either lanthanum and actinium, or lutetium and lawrencium) shown in a single cell of the main table, between barium and hafnium, and radium and rutherfordium, respectively. This convention is entirely a matter of aesthetics and formatting practicality; a rarely used wide-formatted periodic table inserts the lanthanide and actinide series in their proper places, as parts of the table’s sixth and seventh rows (periods).
|
Lanthanides
|
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||
Etymology[edit]
Together with scandium and yttrium, the trivial name “rare earths” is sometimes used to describe all the lanthanides. This name arises from the minerals from which they were isolated, which were uncommon oxide-type minerals. However, the use of the name is deprecated by IUPAC, as the elements are neither rare in abundance nor “earths” (an obsolete term for water-insoluble strongly basic oxides of electropositive metals incapable of being smelted into metal using late 18th century technology)[citation needed]. Cerium is the 26th most abundant element in the Earth’s crust, neodymium is more abundant than gold and even thulium (the least common naturally occurring lanthanide) is more abundant than iodine,[5] which is itself common enough for biology to have evolved critical usages thereof. Despite their abundance, even the technical term “lanthanides” could be interpreted to reflect a sense of elusiveness on the part of these elements, as it comes from the Greek λανθανειν (lanthanein), “to lie hidden”. However, if not referring to their natural abundance, but rather to their property of “hiding” behind each other in minerals, this interpretation is in fact appropriate. The etymology of the term must be sought in the first discovery of lanthanum, at that time a so-called new rare earth element “lying hidden” in a cerium mineral, and it is an irony that lanthanum was later identified as the first in an entire series of chemically similar elements and could give name to the whole series. The term “lanthanide” was introduced by Victor Goldschmidt in 1925.[6]
Physical properties of the elements[edit]
| Chemical element | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Atomic number | 57 | 58 | 59 | 60 | 61 | 62 | 63 | 64 | 65 | 66 | 67 | 68 | 69 | 70 | 71 |
| Image |  |
 |
 |
 |
|||||||||||
| Density (g/cm3) | 6.162 | 6.770 | 6.77 | 7.01 | 7.26 | 7.52 | 5.244 | 7.90 | 8.23 | 8.540 | 8.79 | 9.066 | 9.32 | 6.90 | 9.841 |
| Melting point (°C) | 920 | 795 | 935 | 1024 | 1042 | 1072 | 826 | 1312 | 1356 | 1407 | 1461 | 1529 | 1545 | 824 | 1652 |
| Boiling point (°C) | 3464 | 3443 | 3520 | 3074 | 3000 | 1794 | 1529 | 3273 | 3230 | 2567 | 2720 | 2868 | 1950 | 1196 | 3402 |
| Atomic electron configuration* | 5d1 | 4f15d1 | 4f3 | 4f4 | 4f5 | 4f6 | 4f7 | 4f75d1 | 4f9 | 4f10 | 4f11 | 4f12 | 4f13 | 4f14 | 4f145d1 |
| Metal lattice (RT) | dhcp | fcc | dhcp | dhcp | dhcp | ** | bcc | hcp | hcp | hcp | hcp | hcp | hcp | hcp | hcp |
| Metallic radius pm | 162 | 181.8 | 182.4 | 181.4 | 183.4 | 180.4 | 208.4 | 180.4 | 177.3 | 178.1 | 176.2 | 176.1 | 175.9 | 193.3 | 173.8 |
| Resistivity (25 °C) /μ Ohm cm | 57-80 20 °C |
73 | 68 | 64 | 88 | 90 | 134 | 114 | 57 | 87 | 87 | 79 | 29 | 79 | |
| mag susceptibility χmol /10−6(cm3·mol−1) |
+95.9 | +2500 (β) | +5530(α) | +5930 (α) | +1278(α) | +30900 | +185000 (350 K) |
+170000 (α) | +98000 | +72900 | +48000 | +24700 | +67 (β) | +183 |
* Between initial [Xe] and final 6s2 electronic shells ** Sm has a close packed structure like the other lanthanides but has an unusual 9 layer repeat
Gschneider and Daane (1988) attribute the trend in melting point which increases across the series, (lanthanum (920 °C) – lutetium (1622 °C)) to the extent of hybridisation of the 6s, 5d and 4f orbitals. The hybridisation is believed to be at its greatest for cerium which has the lowest melting point of all, 795 °C.[7] The lanthanide metals are soft, their hardness increases across the series.[8] Europium stands out as it has the lowest density in the series at 5.24 g/cm3 and the largest metallic radius in the series at 208.4 pm. It can be compared to barium which has a metallic radius of 222 pm. It is believed that the metal contains the larger Eu2+ ion and that there are only two electrons in the conduction band. Ytterbium also has large metallic radius and a similar explanation is suggested.[8] The resistivities of the lanthanide metals are relatively high, ranging from 29 to 134 μ Ohm·cm. These values can be compared to a good conductor such as aluminium which has a resistivity of 2.655 μ Ohm·cm. With the exceptions of La, Yb and Lu (which have no unpaired f electrons) the lanthanides are strongly paramagnetic and this is reflected in their magnetic susceptibilities. Gadolinium becomes ferromagnetic at below 16 °C (Curie point). The other heavier lanthanides, terbium, dysprosium, holmium, erbium, thulium and ytterbium become ferromagnetic at much lower temperatures.[9]
Chemistry and compounds[edit]
| Chemical element | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Atomic number | 57 | 58 | 59 | 60 | 61 | 62 | 63 | 64 | 65 | 66 | 67 | 68 | 69 | 70 | 71 |
| Ln3+ electron configuration*[10] | 4f0 | 4f1 | 4f2 | 4f3 | 4f4 | 4f5 | 4f6 | 4f7 | 4f8 | 4f9 | 4f10 | 4f11 | 4f12 | 4f13 | 4f14 |
| Ln3+ radius (pm)[8] | 103 | 102 | 99 | 98.3 | 97 | 95.8 | 94.7 | 93.8 | 92.3 | 91.2 | 90.1 | 89 | 88 | 86.8 | 86.1 |
| Ln4+ ion colour in aqueous solution[11] | — | Orange-yellow | Yellow | Blue-violet | — | — | — | — | Red-brown | Orange-yellow | — | — | — | — | — |
| Ln3+ ion colour in aqueous solution[10] | Colorless | Colorless | Green | Violet | Pink | Pale yellow | Colorless | Colorless | V. pale pink | Pale yellow | Yellow | Rose | Pale green | Colorless | Colorless |
| Ln2+ ion colour in aqueous solution[8] | — | — | — | — | — | Blood red | Colorless | — | — | — | — | — | Violet-red | Yellow-green | — |
* Between initial [Xe] and final 6s2 electronic shells
| Oxidation state | 57 | 58 | 59 | 60 | 61 | 62 | 63 | 64 | 65 | 66 | 67 | 68 | 69 | 70 | 71 |
| +2 | Sm2+ | Eu2+ | Tm2+ | Yb2+ | |||||||||||
| +3 | La3+ | Ce3+ | Pr3+ | Nd3+ | Pm3+ | Sm3+ | Eu3+ | Gd3+ | Tb3+ | Dy3+ | Ho3+ | Er3+ | Tm3+ | Yb3+ | Lu3+ |
| +4 | Ce4+ | Pr4+ | Nd4+ | Tb4+ | Dy4+ |
Effect of 4f orbitals[edit]
Going across the lanthanides, in the periodic table, the 4f orbitals are usually being filled. The effect of the 4f orbitals on the chemistry of the lanthanides is profound and is the factor that distinguishes them from the transition metals. There are seven 4f orbitals and there are two different ways in which they are depicted, firstly as a “cubic set” or as a general set. The cubic set is fz3, fxz2, fyz2, fxyz, fz(x2−y2), fx(x2−3y2) and fy(3x2−y2). The 4f orbitals penetrate the [Xe] core and are isolated and do not participate in bonding. This explains why crystal field effects are small and why they do not form π bonds.[10] As there are seven 4f orbitals the number of unpaired electrons can be as high as 7 which gives rise to the large magnetic moments observed for lanthanide compounds. Measuring the magnetic moment can be used to investigate the 4f electron configuration and this is a useful tool in providing an insight into the chemical bonding.[14] The lanthanide contraction, the reduction in size of the Ln3+ ion from La3+(103 pm)- Lu3+(86.1 pm) is often explained by the poor shielding of the 5s and 5p electrons by the 4f electrons.[10]

Lanthanide oxides: clockwise from top center: praseodymium, cerium, lanthanum, neodymium, samarium and gadolinium.
The electronic structure of the lanthanide elements, with minor exceptions is [Xe]6s24fn. The chemistry of the lanthanides is dominated by the +3 oxidation state and in LnIII compounds the 6s electrons and (usually) one 4f electron are lost and the ions have the configuration [Xe]4fm.[15] All the lanthanide elements exhibit the oxidation state +3. In addition Ce3+ can lose its single f electron to form Ce4+ with the stable electronic configuration of xenon. Also, Eu3+ can gain an electron to form Eu2+ with the f7 configuration which has the extra stability of a half-filled shell. Other than Ce(IV) and Eu(II), none of the lanthanides are stable in oxidation states other than +3 in aqueous solution.Promethium is effectively a man-made element as all its isotopes are radioactive with half-lives shorter than 20 years.
In terms of reduction potentials, the Ln0/3+ couples are nearly the same for all lanthanides, ranging from −1.99 (for Eu) to −2.35 V (for Pr). Thus, these metals are highly reducing, with reducing power similar to alkaline earth metals such as Mg (−2.36 V).[8]
Lanthanide oxidation states[edit]
All of the lanthanide elements are commonly known to have the +3 oxidation state and it was thought that only samarium, europium, and ytterbium had the +2 oxidation readily accessible in solution. Now, it is known that all of the lanthanides can form +2 complexes in solution.[16]
| [show]Chemical element | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
|---|
The ionisation energies for the lanthanides can be compared with aluminium. In aluminium the sum of the first three ionisation energies is 5139 kJ·mol−1, whereas the lanthanides fall in the range 3455 – 4186 kJ·mol−1. This correlates with the highly reactive nature of the lanthanides.
The sum of the first two ionisation energies for europium, 1632 kJ·mol−1 can be compared with that of barium 1468.1 kJ·mol−1 and europium’s third ionisation energy is the highest of the lanthanides. The sum of the first two ionisation energies for ytterbium are the second lowest in the series and its third ionisation energy is the second highest. The high third ionisation energy for Eu and Yb correlate with the half filling 4f7 and complete filling 4f14 of the 4f sub shell, and the stability afforded by such configurations due to exchange energy.[10] Europium and ytterbium form salt like compounds with Eu2+ and Yb2+, for example the salt like dihydrides,.[17] Both europium and ytterbium dissolve in liquid ammonia forming solutions of Ln2+(NH3)x again demonstrating their similarities to the alkaline earth metals.[8]
The relative ease with which the 4th electron can be removed in cerium and (to a lesser extent praseodymium) indicates why Ce(IV) and Pr(IV) compounds can be formed, for example CeO2 is formed rather than Ce2O3 when cerium reacts with oxygen.
Separation of lanthanides[edit]
The similarity in ionic radius between adjacent lanthanide elements makes it difficult to separate them from each other in naturally occurring ores and other mixtures. Historically, the very laborious processes of cascading and fractional crystallization were used. Because the lanthanide ions have slightly different radii, the lattice energy of their salts and hydration energies of the ions will be slightly different, leading to a small difference in solubility. Salts of the formula Ln(NO3)3·2NH4NO3·4H2O can be used. Industrially, the elements are separated from each other by solvent extraction. Typically an aqueous solution of nitrates is extracted into kerosene containing tri-n-butylphosphate. The strength of the complexes formed increases as the ionic radius decreases, so solubility in the organic phase increases. Complete separation can be achieved continuously by use ofcountercurrent exchange methods. The elements can also be separated by ion-exchange chromatography, making use of the fact that the stability constant for formation ofEDTA complexes increases for log K ≈ 15.5 for [La(EDTA)]− to log K ≈ 19.8 for [Lu(EDTA)]−.[8][18]
Coordination chemistry and catalysis[edit]
When in the form of coordination complexes, lanthanides exist overwhelmingly in their +3 oxidation state, although particularly stable 4f configurations can also give +4 (Ce, Tb) or +2 (Eu, Yb) ions. All of these forms are strongly electropositive and thus lanthanide ions are hard Lewis acids. The oxidation states are also very stable and with the exception of SmI2[19] and cerium(IV) salts[20] lanthanides are not used for redox chemistry. 4f electrons have a high probability of being found close to the nucleus and are thus strongly affected as the nuclear charge increases across the series; this results in a corresponding decrease in ionic radii referred to as the lanthanide contraction.
The low probability of the 4f electrons existing at the outer region of the atom or ion permits little effective overlap between the orbitals of a lanthanide ion and any binding ligand. Thus lanthanide complexes typically have little or no covalent character and are not influenced by orbital geometries. The lack of orbital interaction also means that varying the metal typically has little effect on the complex (other than size), especially when compared to transition metals. Complexes are held together by weaker electrostatic forces which are omni-directional and thus the ligands alone dictate the symmetry and coordination of complexes. Steric factors therefore dominate, with coordinative saturation of the metal being balanced against inter-ligand repulsion. This results in a diverse range of coordination geometries, many of which are irregular,[21] and also manifests itself in the highlyfluxional nature of the complexes. As there is no energetic reason to be locked into a single geometry rapid intramolecular and intermolecular ligand exchange will take place, which typically results in complexes which will rapidly fluctuate between all possible configurations.
Many of these features make lanthanide complexes effective catalysts. Hard Lewis acids are able to polarise bonds upon coordination and thus alter the electrophilicity of compounds, with a classic example being the Luche reduction. The large size of the ions coupled with their labile ionic bonding allows even bulky coordinating species to bind and dissociate rapidly, resulting in very high turnover rates; thus excellent yields can often be achieved with loadings of only a few mol%.[22] The lack of orbital interactions combined with the lanthanide contraction means that the lanthanides change in size across the series but that their chemistry remains much the same. This allows for easy tuning of the steric environments and examples exist where this has been used to improve the catalytic activity of the complex[23][24][25] and change the nuclearity of metal clusters.[26][27]
Despite this, the use of lanthanide coordination complexes as homogeneous catalysts is largely restricted to the laboratory and there are currently few examples them being used on an industrial scale.[28] It should be noted however, that lanthanides exist in many forms other that coordination complexes and many of these are industrially useful. In particular lanthanide metal oxides are used as heterogeneous catalysts in various industrial processes.
Ln(III) compounds[edit]
The trivalent lanthanides mostly form ionic salts. The trivalent ions are hard acceptors and form more stable complexes with oxygen-donor ligands than with nitrogen-donor ligands. The larger ions are 9-coordinate in aqueous solution, [Ln(H2O)9]3+ but the smaller ions are 8-coordinate, [Ln(H2O)8]3+. There is some evidence that the later lanthanides have more water molecules in the second coordination sphere.[29] Complexation with monodentate ligands is generally weak because it is difficult to displace water molecules from the first coordination sphere. Stronger complexes are formed with chelating ligands because of the chelate effect, such as the tetra-anion derived from 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA).
Ln(II) and Ln(IV) compounds[edit]
The most common divalent derivatives of the lanthanides are for Eu(II), which achieves a favorable f7 configuration. Divalent halide derivatives are known for all of the lanthanides. They are either conventional salts or are Ln(III) “electride“-like salts. The simple salts include YbI2, EuI2, and SmI2. The electride-like salts, described as Ln3+, 2I−, e−, include LaI2, CeI2 and GdI2. Many of the iodides form soluble complexes with ethers, e.g. TmI2(dimethoxyethane)3.[30] Samarium(II) iodide is a useful reducing agent. Ln(II) complexes can be synthesized by transmetalation reactions.
Ce(IV) in ceric ammonium nitrate is a useful oxidizing agent. Otherwise tetravalent lanthanides are rare. The Ce(IV) is the exception owing to the tendency to form an unfilled f shell.
Hydrides[edit]
| [show]Chemical element | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
|---|
Lanthanide metals react exothermically with hydrogen to form LnH2, dihydrides.[17] With the exception of Eu and Yb which resemble the Ba and Ca hydrides (non conducting,transparent salt like compounds) they form black pyrophoric, conducting compounds[35] where the metal sub-lattice is face centred cubic and the H atoms occupy tetrahedral sites.[17] Further hydrogenation produces a trihydride which is non-stoichiometric, non-conducting, more salt like. The formation of trihydride is associated with and increase in 8-10% volume and this is linked to greater localisation of charge on the hydrogen atoms which become more anionic (H− hydride anion) in character.[17]
Halides[edit]
| [show]Chemical element | La | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu |
|---|
The only tetrahalides known are those of cerium, praseodymium and terbium mirroring the formation of the dioxides.[8] All of the lanthanides form trihalides with fluorine, chlorine, bromine and iodine. They are all high melting and predominantly ionic in nature.[8] The fluorides are only slightly soluble in water and are not sensitive to air, and this contrasts with the other halides which are air sensitive, readily soluble in water and react at high temperature to form oxohalides.[41] The trihalides were important as pure metal can be prepared from them.[8] In the gas phase the trihalides are planar or approximately planar, the lighter lanthanides have a lower % of dimers, the heavier lanthanides a higher proportion. The dimers have a similar structure to Al2Cl6[42]
Some of the dihalides are conducting while the rest are insulators. The conducting forms can be considered as LnIII electride compounds where the electron is delocalised into a conduction band, Ln3+ (X−)2(e−). All of the diodides have relatively short metal-metal separations.[36] The CuTi2 structure of the lanthanum, cerium and praseodymium diodides along with HP-NdI2 contain 44 nets of metal and iodine atoms with short metal-metal bonds (393-386 La-Pr).[36] these compounds should be considered to be two-dimensional metals (two-dimensional in the same way that graphite is). The salt like dihalides include those of Eu Dy Tm and Yb. The formation of a relatively stable +2 oxidation state for Eu and Yb is usually explained by the stability (exchange energy) of half filled (f7) and fully filled f14. GdI2 possesses the layered MoS2 structure, is ferromagnetic and exhibits colossal magnetoresistance[36] The sesquihalides Ln2X3 and the Ln7I12 compounds listed in the table contain metal clusters, discrete Ln6I12 clusters in Ln7I12 and condensed clusters forming chains in the sesquihalides. Scandium forms a similar cluster compound with chlorine, Sc7Cl12[8] Unlike many transition metal clusters these lanthanide clusters do not have strong metal-metal interactions and this is due to the low number of valence electrons involved, but instead are stabilised by the surrounding halogen atoms.[36]
LaI is the only known monohalide. Prepared from the reaction of LaI3 and La metal, it has a NiAs type structure and can be formulated La3+ (I−)(e−)2.[40]
Oxides and hydroxides[edit]
All of the lanthanides form sesquioxides, Ln2O3. The lighter/larger lanthanides adopt a hexagonal 7-coordinate structure while the heavier/smaller ones adopt a cubic 6-coordinate “C-M2O3” structure.[37] All of the sesquioxides are basic, and absorb water and carbon dioxide from air to form carbonates, hydroxides and hydroxycarbonates.[43]They dissolve in acids to form salts.[10]
Cerium forms a stoichiometric dioxide, CeO2, where cerium has an oxidation state of +4. CeO2 is basic and dissolves with difficulty in acid to form Ce4+ solutions, from which CeIVsalts can be isolated, for example the hydrated nitrate Ce(NO3)4.5H2O. CeO2 is used as an oxidation catalyst in catalytic converters.[10] Praseodymium and terbium form non-stoichiometric oxides containing LnIV,[10] although more extreme reaction conditions can produce stoichiometric (or near stoichiometric) PrO2 and TbO2.[8]
Europium and ytterbium form salt-like monoxides, EuO and YbO, which have a rock salt structure.[10] EuO is ferromagnetic at low temperatures,[8] and is a semiconductor with possible applications in spintronics.[44] A mixed EuII/EuIII oxide Eu3O4 can be produced by reducing Eu2O3 in a stream of hydrogen.[43] Neodymium and samarium also form monoxides, but these are shiny conducting solids,[8] although the existence of samarium monoxide is considered dubious.[43]
All of the lanthanides form hydroxides, Ln(OH)3. With the exception of lutetium hydroxide, which has a cubic structure, they have the hexagonal UCl3 structure.[43] The hydroxides can be precipitated from solutions of LnIII.[10] They can also be formed by the reaction of the sesquioxide, Ln2O3, with water, but although this reaction is thermodynamically favourable it is kinetically slow for the heavier members of the series.[43] Fajan’s rules indicate that the smaller Ln3+ ions will be more polarizing and their salts correspondingly less ionic. The hydroxides of the heavier lanthanides become less basic, for example Yb(OH)3 and Lu(OH)3 are still basic hydroxides but will dissolve in hot concentratedNaOH.[8]
Chalcogenides (S, Se, Te)[edit]
All of the lanthanides form Ln2Q3 (Q= S, Se, Te).[10] The sesquisulfides can be produced by reaction of the elements or (with the exception of Eu2S3) sulfidizing the oxide (Ln2O3) with H2S.[10] The sesquisulfides, Ln2S3 generally lose sulfur when heated and can form a range of compositions between Ln2S3 and Ln3S4. The sesquisulfides are insulators but some of the Ln3S4 are metallic conductors (e.g. Ce3S4) formulated (Ln3+)3 (S2−)4 (e−), while others (e.g. Eu3S4 and Sm3S4) are semiconductors.[10] Structurally the sesquisulfides adopt structures that vary according the size of the Ln metal. The lighter and larger lanthanides favouring 7 coordinate metal atoms, the heaviest and smallest lanthanides (Yb and Lu) favouring 6 coordination and the rest structures with a mixture of 6 and 7 coordination.[10] Polymorphism is common amongst the sesquisulfides.[45] The colors of the sesquisulfides vary metal to metal and depend on the polymorphic form. The colors of the γ-sesquisulfides are La2S3, white/yellow; Ce2S3, dark red; Pr2S3, green; Nd2S3, light green; Gd2S3, sand; Tb2S3, light yellow and Dy2S3, orange.[46] The shade of γ-Ce2S3 can be varied by doping with Na or Ca with hues ranging from dark red to yellow,[36][46] and Ce2S3 based pigments are used commercially and are seen as low toxicity substitutes for cadmium based pigments.[46]
All of the lanthanides form monochalcogenides, LnQ, (Q= S, Se, Te).[10] The majority of the monochalcogenides are conducting, indicating a formulation LnIIIQ2−(e-) where the electron is in conduction bands. The exceptions are SmQ, EuQ and YbQ which are semiconductors or insulators but exhibit a pressure induced transition to a conducting state.[45]Compounds LnQ2 are known but these do not contain LnIV but are LnIII compounds containing polychalcogenide anions.[47]
Oxysulfides Ln2O2S are well known, they all have the same structure with 7 coordinate Ln atoms with 3 sulfur atoms and 4 oxygen as near neighbours.[48] Doping these with other lanthanide elements produces phosphors. As an example, gadolinium oxysulfide, Gd2O2S doped with Tb3+ produces visible photons when irradiated with high energy X-rays and is used as a scintillator in flat panel detectors.[49] When mischmetal, an alloy of lanthanide metals, is added to molten steel to remove oxygen and sulfur, stable oxysulfides are produced that form an immiscible solid.[10]
Pnictides (group 15)[edit]
All of the lanthanides form a mononitride, LnN, with the rock salt structure. The mononitrides have attracted interest because of their unusual physical properties. SmN and EuN are reported as being “half metals“.[36] NdN, GdN, TbN and DyN are ferromagnetic, SmN is antiferromagnetic.[50] Applications in the field of spintronics are being investigated.[44]CeN is unusual as it is a metallic conductor, contrasting with the other nitrides also with the other cerium pnictides. A simple description is Ce4+ N3− (e–) but the interatomic distances are a better match for the trivalent state rather than for the tetravalent state. A number of different explanations have been offered.[51] The nitrides can be prepared by the reaction of lanthanum metals with nitrogen. Some nitride is produced along with the oxide, when lanthanum metals are ignited in air.[52] Alternative methods of synthesis are a high temperature reaction of lanthanide metals with ammonia or the decomposition of lanthanide amides, Ln(NH2)3. Achieving pure stoichiometric compounds, and crystals with low defect density has proved difficult.[44] The lanthanide nitrides are sensitive to air and hydrolyse producing ammonia.[35]
The other pnictides phosphorus, arsenic, antimony and bismuth also react with the lanthanide metals to form monopnictides, LnQ. Additionally a range of other compounds can be produced with varying stoichiometries, such as LnP2, LnP5, LnP7 Ln3As, Ln5As3 and LnAs2.[53]
Carbides[edit]
Carbides of varying stoichiometries are known for the lanthanides. Non-stoichiometry is common. All of the lanthanides form LnC2 and Ln2C3 which both contain C2 units. The dicarbides with exception of EuC2, are metallic conductors with the calcium carbide structure and can be formulated as Ln3+C22−(e–). The C-C bond length is longer than that inCaC2, which contains the C22− anion, indicating that the antibonding orbitals of the C22− anion are involved in the conduction band. These dicarbides hydrolyse to form hydrogen and a mixture of hydrocarbons.[54] EuC2 and to a lesser extent YbC2 hydrolyse differently producing a higher percentage of acetylene (ethyne).[55] The sesquicarbides, Ln2C3 can be formulated as Ln4(C2)3. These compounds adopt the Pu2C3 structure[36] which has been described as having C22− anions in bisphenoid holes formed by eight near Ln neighbours.[56] The lengthening of the C-C bond is less marked in the sesquicarbides than in the dicarbides, with the exception of Ce2C3.[54] Other carbon rich stoichiometries are known for some lanthanides. Ln3C4 (Ho-Lu) containing C, C2 and C3 units;[57] Ln4C7 (Ho- Lu) contain C atoms and C3 units[58] and Ln4C5 (Gd-Ho) containing C and C2 units.[59]Metal rich carbides contain interstitial C atoms and no C2 or C3 units. These are Ln4C3 (Tb and Lu); Ln2C (Dy, Ho, Tm)[60][61] and Ln3C[36] (Sm-Lu).
Borides[edit]
All of the lanthanides form a number of borides. The “higher” borides (LnBx where x > 12) are insulators/semiconductors whereas the lower borides are typically conducting. The lower borides have stoichiometries of LnB2, LnB4, LnB6 and LnB12.[62] Applications in the field of spintronics are being investigated.[44] The range of borides formed by the lanthanides can be compared to those formed by the transition metals. The boron rich borides are typical of the lanthanides (and groups 1-3) whereas for the transition metals tend to form metal rich, “lower” borides.[63] The lanthanide borides are typically grouped together with the group 3 metals with which they share many similarities of reactivity, stoichiometry and structure. Collectively these are then termed the rare earth borides.[62]
Many methods of producing lanthanide borides have been used, amongst them are direct reaction of the elements; the reduction of Ln2O3 with boron; reduction of boron oxide, B2O3, and Ln2O3 together with carbon; reduction of metal oxide with boron carbide, B4C.[62][63][64][65] Producing high purity samples has proved to be difficult.[65] Single crystals of the higher borides have been grown in a low melting metal (e.g. Sn, Cu, Al).[62]
Diborides, LnB2, have been reported for Sm, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu. All have the same, AlB2, structure containing a graphitic layer of boron atoms. Low temperature ferromagnetic transitions for Tb, Dy, Ho and Er. TmB2 is ferromagnetic at 7.2 K.[36]
Tetraborides, LnB4 have been reported for all of the lanthanides except EuB4, all have the same UB4 structure. The structure has a boron sub-lattice consists of chains of octahedral B6 clusters linked by boron atoms. The unit cell decreases in size successively from LaB4 to LuB4. The tetraborides of the lighter lanthanides melt with decomposition to LnB6.[65] Attempts to make EuB4 have failed.[64] The LnB4 are good conductors[62] and typically antiferromagnetic.[36]
Hexaborides, LnB6 have been reported for all of the lanthanides. They all have the CaB6 structure, containing B6 clusters. They are non-stoichiometric due to cation defects. The hexaborides of the lighter lanthanides (La – Sm) melt without decomposition, EuB6 decomposes to boron and metal and the heavier lanthanides decompose to LnB4 with exception of YbB6 which decomposes forming YbB12. The stability has in part been correlated to differences in volatility between the lanthanide metals.[65] In EuB6 and YbB6 the metals have an oxidation state of +2 whereas in the rest of the lanthanide hexaborides it is +3. This rationalises the differences in conductivity, the extra electrons in the LnIIIhexaborides entering conduction bands. EuB6 is a semiconductor and the rest are good conductors.[36][65] LaB6 and CeB6 are thermionic emitters, used, for example, in scanning electron microscopes.[66]
Dodecaborides, LnB12, are formed by the heavier smaller lanthanides, but not by the lighter larger metals, La – Eu. With the exception YbB12 (where Yb takes an intermediate valence and is a Kondo insulator), the dodecaborides are all metallic compounds. They all have the UB12 structure containing a 3 dimensional framework of cubooctahedral B12clusters.[62]
The higher boride LnB66 is known for all lanthanide metals. The composition is approximate as the compounds are non-stoichiometric.[62] They all have similar complex structurewith over 1600 atoms in the unit cell. The boron cubic sub lattice contains super icosahedra made up of a central B12 icosahedra surrounded by 12 others, B12(B12)12.[62] Other complex higher borides LnB50 (Tb, Dy, Ho Er Tm Lu) and LnB25 are known (Gd, Tb, Dy, Ho, Er) and these contain boron icosahedra in the boron framework.[62]
Organometallic compounds[edit]
Lanthanide-carbon σ bonds are well known; however as the 4f electrons have a low probability of existing at the outer region of the atom there is little effective orbital overlap, resulting in bonds with significant ionic character. As such organo-lanthanide compounds exhibit carbanion-like behaviour, unlike in transition metal organometallic compounds. Because of their large size, lanthanides tend to form more stable organometallic derivatives with bulky ligands to give compounds such as Ln[CH(SiMe3)3].[67] Similarly complexes of cyclopentadienyl anion (Cp−), e.g. [Ln(C5H5)3], are far less common than the corresponding pentamethylcyclopentadienyl, e.g. [Ln(C5Me5)3Cl]. Analogues of uranocene are derived from dilithiocyclooctatetraene, Li2C8H8. Organic lanthanide(II) compounds are also known, such as Cp*2Eu.[30]
Physical properties[edit]
Magnetic and spectroscopic[edit]
All the trivalent lanthanide ions, except lanthanum and lutetium, have unpaired f electrons. However, the magnetic moments deviate considerably from the spin-only valuesbecause of strong spin-orbit coupling. The maximum number of unpaired electrons is 7, in Gd3+, with a magnetic moment of 7.94 B.M., but the largest magnetic moments, at 10.4–10.7 B.M., are exhibited by Dy3+ and Ho3+. However, in Gd3+ all the electrons have parallel spin and this property is important for the use of gadolinium complexes ascontrast reagent in MRI scans.

A solution of 4% holmium oxide in 10% perchloric acid, permanently fused into a quartz cuvette as a wavelength calibration standard
Crystal field splitting is rather small for the lanthanide ions and is less important than spin-orbit coupling in regard to energy levels.[8]Transitions of electrons between f orbitals are forbidden by the Laporte rule. Furthermore, because of the “buried” nature of the f orbitals, coupling with molecular vibrations is weak. Consequently, the spectra of lanthanide ions are rather weak and the absorption bands are similarly narrow. Glass containing holmium oxide and holmium oxide solutions (usually in perchloric acid) have sharp optical absorption peaks in the spectral range 200–900 nm and can be used as a wavelength calibration standard for optical spectrophotometers,[68] and are available commercially.[69]
As f-f transitions are Laporte-forbidden, once an electron has been excited, decay to the ground state will be slow. This makes them suitable for use in lasers as it makes the population inversion easy to achieve. The Nd:YAG laser is one that is widely used. Europium-doped yttrium vanadate was the first red phosphor to enable the development of color television screens.[70] Lanthanide ions have notable luminescent properties due to their unique 4f orbitals. Laporte forbidden f-f transitions can be activated by excitation of a bound “antenna” ligand. This leads to sharp emission bands throughout the visible, NIR, and IR and relatively long luminescence lifetimes.[71]
Occurrence[edit]
Main article: Rare earth element § Geological distribution
The lanthanide contraction is responsible for the great geochemical divide that splits the lanthanides into light and heavy-lanthanide enriched minerals, the latter being almost inevitably associated with and dominated by yttrium. This divide is reflected in the first two “rare earths” that were discovered: yttria (1794) and ceria (1803). The geochemical divide has put more of the light lanthanides in the Earth’s crust, but more of the heavy members in the Earth’s mantle. The result is that although large rich ore-bodies are found that are enriched in the light lanthanides, correspondingly large ore-bodies for the heavy members are few. The principal ores are monazite and bastnäsite. Monazite sands usually contain all the lanthanide elements, but the heavier elements are lacking in bastnäsite. The lanthanides obey the Oddo-Harkins rule – odd-numbered elements are less abundant than their even-numbered neighbors.
Three of the lanthanide elements have radioactive isotopes with long half-lives (138La, 147Sm and 176Lu) that can be used to date minerals and rocks from Earth, the Moon and meteorites.[72]
Applications[edit]
Industrial[edit]
Lanthanide elements and their compounds have many uses but the quantities consumed are relatively small in comparison to other elements. About 15000 ton/year of the lanthanides are consumed as catalysts and in the production of glasses. This 15000 tons corresponds to about 85% of the lanthanide production. From the perspective of value, however, applications in phosphors and magnets are more important.[73]
The devices lanthanide elements are used in include superconductors, samarium-cobalt and neodymium-iron-boron high-flux rare-earth magnets, magnesium alloys, electronic polishers, refining catalysts and hybrid car components (primarily batteries and magnets).[74] Lanthanide ions are used as the active ions in luminescent materials used inoptoelectronics applications, most notably the Nd:YAG laser. Erbium-doped fiber amplifiers are significant devices in optical-fiber communication systems. Phosphors with lanthanide dopants are also widely used in cathode ray tube technology such as television sets. The earliest color television CRTs had a poor-quality red; europium as a phosphor dopant made good red phosphors possible. Yttrium iron garnet (YIG) spheres can act as tunable microwave resonators. Lanthanide oxides are mixed with tungsten to improve their high temperature properties for welding, replacing thorium, which was mildly hazardous to work with. Many defense-related products also use lanthanide elements such as night vision goggles and rangefinders. The SPY-1 radar used in some Aegis equipped warships, and the hybrid propulsion system of Arleigh Burke-class destroyers all use rare earth magnets in critical capacities.[75] The price for lanthanum oxide used in fluid catalytic cracking has risen from $5 per kilogram in early 2010 to $140 per kilogram in June 2011.[76]
Most lanthanides are widely used in lasers, and as (co-)dopants in doped-fiber optical amplifiers; for example, in Er-doped fiber amplifiers, which are used as repeaters in the terrestrial and submarine fiber-optic transmission links that carry internet traffic. These elements deflect ultraviolet and infrared radiation and are commonly used in the production of sunglass lenses. Other applications are summarized in the following table:[5]
| Application | Percentage |
|---|---|
| Catalytic converters | 45 |
| Petroleum refining catalysts | 25 |
| Permanent magnets | 12 |
| Glass polishing and ceramics | 7 |
| Metallurgical | 7 |
| Phosphors | 3 |
| Other | 1 |
The complex Gd(DOTA) is used in magnetic resonance imaging.
Life science[edit]
As mentioned in the industrial applications section above, lanthanide metals are particularly useful in technologies that take advantage of their reactivity to specific wavelengths of light.[77] Certain life science applications take advantage of the unique fluorescence properties of lanthanide ion complexes (Ln(III) chelates or cryptates). These are well-suited for this application due to their large Stokes shifts and extremely long emission lifetimes (from microseconds to milliseconds) compared to more traditional fluorophores (e.g.,fluorescein, allophycocyanin, phycoerythrin, and rhodamine). The biological fluids or serum commonly used in these research applications contain many compounds and proteins which are naturally fluorescent. Therefore, the use of conventional, steady-state fluorescence measurement presents serious limitations in assay sensitivity. Long-lived fluorophores, such as lanthanides, combined with time-resolved detection (a delay between excitation and emission detection) minimizes prompt fluorescence interference.
Time-resolved fluorometry (TRF) combined with fluorescence resonance energy transfer (FRET) offers a powerful tool for drug discovery researchers: Time-Resolved Fluorescence Resonance Energy Transfer or TR-FRET. TR-FRET combines the low background aspect of TRF with the homogeneous assay format of FRET. The resulting assay provides an increase in flexibility, reliability and sensitivity in addition to higher throughput and fewer false positive/false negative results.
This method involves two fluorophores: a donor and an acceptor. Excitation of the donor fluorophore (in this case, the lanthanide ion complex) by an energy source (e.g. flash lamp or laser) produces an energy transfer to the acceptor fluorophore if they are within a given proximity to each other (known as the Förster’s radius). The acceptor fluorophore in turn emits light at its characteristic wavelength.
The two most commonly used lanthanides in life science assays are shown below along with their corresponding acceptor dye as well as their excitation and emission wavelengths and resultant Stokes shift (separation of excitation and emission wavelengths).
| Donor | Excitation⇒Emission λ (nm) | Acceptor | Excitation⇒Emission λ (nm) | Stoke’s Shift (nm) |
|---|---|---|---|---|
| Eu3+ | 340⇒615 | Allophycocyanin | 615⇒660 | 320 |
| Tb3+ | 340⇒545 | Phycoerythrin | 545⇒575 | 235 |
Biological effects[edit]
Due to their sparse distribution in the earth’s crust and low aqueous solubility, the lanthanides have a low availability in the biosphere, and are not known to naturally form part of any biological molecules. Compared to most other nondietary elements, non-radioactive lanthanides are classified as having low toxicity.[73]
Actinide
From Wikipedia, the free encyclopedia
| Actinides in the periodic table | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

The atomic bomb dropped on Nagasaki had a plutonium charge.[1]
The actinide /ˈæktɨnaɪd/ or actinoid /ˈæktɨnɔɪd/ (IUPAC nomenclature) series encompasses the 15 metallic chemical elements with atomic numbers from 89 to 103, actinium through lawrencium.[2][3][4][5]
The actinide series derives its name from the first element in the series, actinium. The informal chemical symbol An is used in general discussions of actinide chemistry to refer to any actinide. All but one of the actinides are f-block elements, corresponding to the filling of the 5f electron shell; lawrencium, a d-block element, is also generally considered an actinide. In comparison with the lanthanides, also mostlyf-block elements, the actinides show much more variable valence. They all have very large atomic and ionic radii and exhibit an unusually large range of physical properties. While actinium and the late actinides (from americium onwards) behave similarly to the lanthanides, the elements thorium through neptunium are much more similar to transition metals in their chemistry.
Of the actinides, primordial thorium and uranium occur naturally in substantial quantities and small amounts of persisting natural plutoniumhave also been identified. The radioactive decay of uranium produces transient amounts of actinium and protactinium, and atoms ofneptunium and plutonium are occasionally produced from transmutation reactions in uranium ores. The other actinides are purelysynthetic elements.[2][6] Nuclear weapons tests have released at least six actinides heavier than plutonium into the environment; analysis of debris from a 1952 hydrogen bomb explosion showed the presence of americium, curium, berkelium, californium, einsteinium andfermium.[7]
All actinides are radioactive and release energy upon radioactive decay; naturally occurring uranium and thorium, and synthetically produced plutonium are the most abundant actinides on Earth. These are used in nuclear reactors and nuclear weapons. Uranium and thorium also have diverse current or historical uses, and americium is used in the ionization chambers of most modern smoke detectors.
In presentations of the periodic table, the lanthanides and the actinides are customarily shown as two additional rows below the main body of the table,[2] with placeholders or else a selected single element of each series (either lanthanum or lutetium, and either actinium orlawrencium, respectively) shown in a single cell of the main table, between barium and hafnium, and radium and rutherfordium, respectively. This convention is entirely a matter of aesthetics and formatting practicality; a rarely used wide-formatted periodic table inserts the lanthanide and actinide series in their proper places, as parts of the table’s sixth and seventh rows (periods).
|
Actinides
|
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||
Discovery, isolation and synthesis[edit]
| Element | Year | Method |
|---|---|---|
| Neptunium | 1940 | Bombarding 238U by neutrons |
| Plutonium | 1941 | Bombarding 238U by deuterons |
| Americium | 1944 | Bombarding 239Pu by neutrons |
| Curium | 1944 | Bombarding 239Pu by α-particles |
| Berkelium | 1949 | Bombarding 241Am by α-particles |
| Californium | 1950 | Bombarding 242Cm by α-particles |
| Einsteinium | 1952 | As a product of nuclear explosion |
| Fermium | 1952 | As a product of nuclear explosion |
| Mendelevium | 1955 | Bombarding 253Es by α-particles |
| Nobelium | 1965 | Bombarding 243Am by 15N or 238U with α-particles |
| Lawrencium | 1961–1971 | Bombarding 252Cf by 10B or 11B and of 243Am with 18O |
Like the lanthanides, the actinides form a family of elements with similar properties. Within the actinides, there are two overlapping groups: transuranium elements, which follow uranium in the periodic table—andtransplutonium elements, which follow plutonium. Compared to the lanthanides, which (except for promethium) are found in nature in appreciable quantities, most actinides are rare. The most abundant, or easy to synthesize actinides are uranium and thorium, followed by plutonium, americium, actinium, protactinium and neptunium.[10]
The existence of transuranium elements was suggested by Enrico Fermi based on his experiments in 1934.[11][12] However, even though four actinides were known by that time, it was not yet understood that they formed a family similar to lanthanides. The prevailing view that dominated early research into transuranics was that they were regular elements in the 7th period, with thorium, protactinium and uranium corresponding to 6th-period hafnium, tantalum and tungsten, respectively. Synthesis of transuranics gradually undermined this point of view. By 1944 an observation that curium failed to exhibit oxidation states above 4 (whereas its supposed 6th period homolog, platinum, can reach oxidation state of 6) prompted Glenn Seaborg to formulate a so-called “actinide hypothesis”. Studies of known actinides and discoveries of further transuranic elements provided more data in support of this point of view, but the phrase “actinide hypothesis” (the implication being that “hypothesis” is something that has not been decisively proven) remained in active use by scientists through the late 1950s.[13][14]
At present, there are two major methods of producing isotopes of transplutonium elements: irradiation of the lighter elements with either neutrons or accelerated charged particles. The first method is most important for applications, as only neutron irradiation using nuclear reactors allows the production of sizeable amounts of synthetic actinides; however, it is limited to relatively light elements. The advantage of the second method is that elements heavier than plutonium, as well as neutron-deficient isotopes, can be obtained, which are not formed during neutron irradiation.[15]
In 1962–1966, there were attempts in the United States to produce transplutonium isotopes using a series of six underground nuclear explosions. Small samples of rock were extracted from the blast area immediately after the test to study the explosion products, but no isotopes with mass number greater than 257 could be detected, despite predictions that such isotopes would have relatively long half-lives of α-decay. This inobservation was attributed to spontaneous fission owing to the large speed of the products and to other decay channels, such as neutron emission and nuclear fission.[16]
From actinium to uranium[edit]

Enrico Fermi suggested the existence of transuranium elements in 1934.
Uranium and thorium were the first actinides discovered. Uranium was identified in 1789 by the German chemist Martin Heinrich Klaprothin pitchblende ore. He named it after the planet Uranus,[6] which had been discovered only eight years earlier. Klaproth was able to precipitate a yellow compound (likely sodium diuranate) by dissolving pitchblende in nitric acid and neutralizing the solution with sodium hydroxide. He then reduced the obtained yellow powder with charcoal, and extracted a black substance that he mistook for metal.[17] Only 60 years later, the French scientist Eugène-Melchior Péligot identified it with uranium oxide. He also isolated the first sample of uranium metal by heating uranium tetrachloride with potassium.[18] The atomic mass of uranium was then calculated as 120, but Dmitri Mendeleevin 1872 corrected it to 240 using his periodicity laws. This value was confirmed experimentally in 1882 by K. Zimmerman.[19][20]
Thorium oxide was discovered by Friedrich Wöhler in the mineral, which was found in Norway (1827).[21] Jöns Jacob Berzeliuscharacterized this material in more detail by in 1828. By reduction of thorium tetrachloride with potassium, he isolated the metal and named it thorium after the Norse god of thunder and lightning Thor.[22][23] The same isolation method was later used by Péligot for uranium.[6]
Actinium was discovered in 1899 by André-Louis Debierne, an assistant of Marie Curie, in the pitchblende waste left after removal of radium and polonium. He described the substance (in 1899) as similar to titanium[24] and (in 1900) as similar to thorium.[25] The discovery of actinium by Debierne was however questioned in 1971[26] and 2000,[27] arguing that Debierne’s publications in 1904 contradicted his earlier work of 1899–1900. The name actinium comes from the Greek aktis, aktinos (ακτίς, ακτίνος), meaning beam or ray. This metal was discovered not by its own radiation but by the radiation of the daughter products.[28][29] Owing to the close similarity of actinium and lanthanum and low abundance, pure actinium could only be produced in 1950. The term actinide was probably introduced by Victor Goldschmidt in 1937.[30][31]
Protactinium was possibly isolated in 1900 by William Crookes.[32] It was first identified in 1913, when Kasimir Fajans and Oswald Helmuth Göhring encountered the short-lived isotope 234mPa (half-life 1.17 minutes) during their studies of the 238U decay. They named the new element brevium (from Latin brevis meaning brief);[33][34] the name was changed to protoactinium (from Greek πρῶτος + ἀκτίς meaning “first beam element”) in 1918 when two groups of scientists, led by the Austrian Lise Meitner and Otto Hahn of Germany and Frederick Soddy and John Cranston of Great Britain, independently discovered 231Pa. The name was shortened to protactinium in 1949. This element was little characterized until 1960, when A. G. Maddock and his co-workers in the U.K. produced 130 grams of protactinium from 60 tonnes of waste left after extraction of uranium from its ore.[35]
Neptunium and above[edit]
Neptunium (named for the planet Neptune, the next planet out from Uranus, after which uranium was named) was discovered by Edwin McMillan and Philip H. Abelson in 1940 inBerkeley, California.[36] They produced the 239Np isotope (half-life = 2.4 days) by bombarding uranium with slow neutrons.[35] It was the first transuranium element produced synthetically.[37]

Glenn T. Seaborg and his group at the University of California at Berkeleysynthesized Pu, Am, Cm, Bk, Cf, Es, Fm, Md, No and element 106, which was later named seaborgium in his honor while he was still living. They also synthesized more than 100 atomic actinide isotopes.
Transuranium elements do not occur in sizeable quantities in nature and are commonly synthesized via nuclear reactions conducted with nuclear reactors. For example, under irradiation with reactor neutrons, uranium-238 partially converts to plutonium-239:
![\mathrm{ {}^{238}_{92}U + {}^{1}_{0}n\ \xrightarrow\ \ {}^{239}_{92}U\ \xrightarrow[23.5\ min]{\beta^-}\ {}^{239}_{93}Np\ \xrightarrow[2.3\ days]{\beta^-}\ {}^{239}_{94}Pu\ \xrightarrow[2.4\cdot 10^4\ years]{\alpha} }](https://upload.wikimedia.org/math/c/1/9/c19737c099a2dc15217a5517fb110620.png)
In this way, Enrico Fermi with collaborators, using the first nuclear reactor Chicago Pile-1, obtained significant amounts of plutonium-239, which were then used in nuclear weapons.[38]
Actinides with the highest mass numbers are synthesized by bombarding uranium, plutonium, curium and californium with ions of nitrogen, oxygen, carbon, neon or boron in a particle accelerator. So, nobelium was produced by bombarding uranium-238 with neon-22 as
 .
.
The first isotopes of transplutonium elements, americium-241 and curium-242, were synthesized in 1944 by Glenn T. Seaborg, Ralph A. James and Albert Ghiorso.[39] Curium-242 was obtained by bombarding plutonium-239 with 32-MeV α-particles
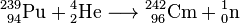 .
.
The americium-241 and curium-242 isotopes also were produced by irradiating plutonium in a nuclear reactor. The latter element was named after Marie Curie and her husband Pierre who are noted for discovering radium and for their work in radioactivity.[40]
Bombarding curium-242 with α-particles resulted in an isotope of californium 245Cf (1950), and a similar procedure yielded in 1949berkelium-243 from americium-241.[41] The new elements were named after Berkeley, California, by analogy with its lanthanidehomologue terbium, which was named after the village of Ytterby in Sweden.[42]
In 1945, B. B. Cunningham obtained the first bulk chemical compound of a transplutonium element, namely americium hydroxide.[43] Over the next three to four years, milligram quantities of americium and microgram amounts of curium were accumulated that allowed production of isotopes of berkelium (Thomson, 1949)[44][45] and californium (Thomson, 1950).[46][47][48] Sizeable amounts of these elements were produced only in 1958 (Burris B. Cunningham and Stanley G. Thomson),[49]and the first californium compound (0.3 µg of CfOCl) was obtained only in 1960 by B. B. Cunningham and J. C. Wallmann.[50]
Einsteinium and fermium were identified in 1952–1953 in the fallout from the “Ivy Mike” nuclear test (1 November 1952), the first successful test of a hydrogen bomb. Instantaneous exposure of uranium-238 to a large neutron flux resulting from the explosion produced heavy isotopes of uranium, including uranium-253 and uranium-255, and their β-decay yielded einsteinium-253 and fermium-255. The discovery of the new elements and the new data on neutron capture were initially kept secret on the orders of the U.S. military until 1955 due to Cold War tensions.[7][51] Nevertheless, the Berkeley team were able to prepare einsteinium and fermium by civilian means, through the neutron bombardment of plutonium-239, and published this work in 1954 with the disclaimer that it was not the first studies that had been carried out on the elements.[52][53] The “Ivy Mike” studies were declassified and published in 1955.[51] The first significant (submicrograms) amounts of einsteinium were produced in 1961 by Cunningham and colleagues, but this has not been done for fermium yet.[54]
The first isotope of mendelevium, 256Md (half-life 87 min), was synthesized by Albert Ghiorso, Glenn T. Seaborg, Gregory R. Choppin, Bernard G. Harvey and Stanley G. Thompson when they bombarded an 253Es target with alpha particles in the 60-inch cyclotron of Berkeley Radiation Laboratory; this was the first isotope of any element to be synthesized one atom at a time.[55]
There were several attempts to obtain isotopes of nobelium by Swedish (1957) and American (1958) groups, but the first reliable result was the synthesis of 256No by the Russian group (Georgy Flyorov et al.) in 1965, as acknowledged by the IUPAC in 1992. In their experiments, Flyorov et al. bombarded uranium-238 with neon-22.[8]
In 1961, Ghiorso et al. obtained the first isotope of lawrencium by irradiating californium (mostly californium-252) with boron-10 and boron-11 ions.[8] The mass number of this isotope was not clearly established (possibly 258 or 259) at the time. In 1965, 256Lr was synthesized by Flyorov et al. from 243Am and 18O. Thus IUPAC recognized the nuclear physics teams at Dubna and Berkeley as the co-discoverers of lawrencium.
Isotopes[edit]
| [show]Isotope | Half-life | Probability of spontaneous fission in % |
Emission energy, MeV (yield in %) |
Specific activity[59] of | ||
|---|---|---|---|---|---|---|

Actinides have 89 to 103 protons and usually have 117 to 159 neutrons.
Thirty-one isotopes of actinium and eight excited isomeric states of some of its nuclides were identified by 2010.[56] Three isotopes, 225Ac,227Ac and 228Ac, were found in nature and the others were produced in the laboratory; only the three natural isotopes are used in applications. Actinium-225 is a member of radioactive neptunium series;[60] it was first discovered in 1947 as a fission product of uranium-233, it is an α-emitter with a half-life of 10 days. Actinium-225 is less available than actinium-228, but is more promising in radiotracer applications.[29] Actinium-227 (half-life 21.77 years) occurs in all uranium ores, but in small quantities. One gram of uranium (in radioactive equilibrium) contains only 2×10−10 gram of 227Ac.[29][56] Actinium-228 is a member of radioactive thorium series formed by the decay of228Ra;[60] it is a β− emitter with a half-life of 6.15 hours. In one tonne of thorium there is 5×10−8 gram of 228Ac. It was discovered by Otto Hahn in 1906.[29]
Twenty nine isotopes of protactinium are known with mass numbers 212–240[56] as well as three excited isomeric states. Only 231Pa and234Pa have been found in nature. All the isotopes have short lifetime, except for protactinium-231 (half-life 32,760 years). The most important isotopes are 231Pa and 233Pa, which is an intermediate product in obtaining uranium-233 and is the most affordable among artificial isotopes of protactinium. 233Pa has convenient half-life and energy of γ-radiation, and thus was used in most studies of protactinium chemistry. Protactinium-233 is a β-emitter with a half-life of 26.97 days.[56][61]
Uranium has the highest number (25) of both natural and synthetic isotopes. They have mass numbers of 217–242,[57] and three of them,234U, 235U and 238U, are present in appreciable quantities in nature. Among others, the most important is 233U, which is a final product of transformations of 232Th irradiated by slow neutrons. 233U has a very higher fission efficiency by low-energy (thermal) neutrons, compared e.g. with 235U. Most uranium chemistry studies were carried out on uranium-238 owing to its long half-life of 4.4×109 years.[62]
There are 19 isotopes of neptunium with mass numbers from 225 to 244;[57] they are all highly radioactive. The most popular among scientists are long-lived 237Np (t1/2 = 2.20×106 years) and short-lived 239Np, 238Np (t1/2 ~ 2 days).[37]
Sixteen isotopes of americium are known with mass numbers from 232 to 248.[57] The most important are 241Am and 243Am, which are alpha-emitters and also emit soft, but intense γ-rays; both of them can be obtained in an isotopically pure form. Chemical properties of americium were first studied with 241Am, but later shifted to 243Am, which is almost 20 times less radioactive. The disadvantage of 243Am is production of the short-lived daughter isotope 239Np, which has to be considered in the data analysis.[63]
Among 19 isotopes of curium,[57] the most accessible are 242Cm and 244Cm; they are α-emitters, but with much shorter lifetime than the americium isotopes. These isotopes emit almost no γ-radiation, but undergo spontaneous fission with the associated emission of neutrons. More long-lived isotopes of curium (245–248Cm, all α-emitters) are formed as a mixture during neutron irradiation of plutonium or americium. Upon short irradiation, this mixture is dominated by curium-246, and then curium-248 begins to accumulate. Both of these isotopes, especially 248Cm, have a longer half-life (3.48×105 years) and are much more convenient for carrying out chemical research than 242Cm and 244Cm, but they also have a rather high rate of spontaneous fission. 247Cm has the longest lifetime among isotopes of curium (1.56×107 years), but is not formed in large quantities because of the strong fission induced by thermal neutrons.
Fourteen isotopes of berkelium were identified with mass numbers 238–252.[57] Only 249Bk is available in large quantities; it has a relatively short half-life of 330 days and emits mostly soft β-particles, which are inconvenient for detection. Its alpha radiation is rather weak (1.45×10−3% with respect to β-radiation), but is sometimes used to detect this isotope. 247Bk is an alpha-emitter with a long half-life of 1,380 years, but it is hard to obtain in appreciable quantities; it is not formed upon neutron irradiation of plutonium because of the β-stability of isotopes of curium isotopes with mass number below 248.[63]
Isotopes of californium with mass numbers 237–256 are formed in nuclear reactors;[57] californium-253 is a β-emitter and the rest are α-emitters. The isotopes with even mass numbers (250Cf, 252Cf and 254Cf) have a high rate of spontaneous fission, especially 254Cf of which 99.7% decays by spontaneous fission. Californium-249 has a relatively long half-life (352 years), weak spontaneous fission and strong γ-emission that facilitates its identification. 249Cf is not formed in large quantities in a nuclear reactor because of the slow β-decay of the parent isotope 249Bk and a large cross section of interaction with neutrons, but it can be accumulated in the isotopically pure form as the β-decay product of (pre-selected) 249Bk. Californium produced by reactor-irradiation of plutonium mostly consists of 250Cf and 252Cf, the latter being predominant for large neutron fluences, and its study is hindered by the strong neutron radiation.[64]
| Parent isotope |
t1/2 | Daughter isotope |
t1/2 | Time to establish radioactive equilibrium |
|---|---|---|---|---|
| 243Am | 7370 years | 239Np | 2.35 days | 47.3 days |
| 245Cm | 8265 years | 241Pu | 14 years | 129 years |
| 247Cm | 1.64×107 years | 243Pu | 4.95 hours | 7.2 days |
| 254Es | 270 days | 250Bk | 3.2 hours | 35.2 hours |
| 255Es | 39.8 days | 255Fm | 22 hours | 5 days |
| 257Fm | 79 days | 253Cf | 17.6 days | 49 days |
Among the 16 known isotopes of einsteinium with mass numbers from 241 to 257[57] the most affordable is 253Es. It is an α-emitter with a half-life of 20.47 days, a relatively weak γ-emission and small spontaneous fission rate as compared with the isotopes of californium. Prolonged neutron irradiation also produces a long-lived isotope 254Es (t1/2 = 275.5 days).[64]
Nineteen isotopes of fermium are known with mass numbers of 242–260. 254Fm, 255Fm and256Fm are α-emitters with a short half-life (hours), which can be isolated in significant amounts.257Fm (t1/2 = 100 days) can accumulate upon prolonged and strong irradiation. All these isotopes are characterized by high rates of spontaneous fission.[64][66]
Among the 15 known isotopes of mendelevium (mass numbers from 245 to 260),[57] the most studied is 256Md, which mainly decays through the electron capture (α-radiation is ≈10%) with the half-life of 77 minutes. Another alpha emitter, 258Md, has a half-life of 53 days. Both these isotopes are produced from rare einsteinium (253Es and 255Es respectively), that therefore limits their availability.[56]
Long-lived isotopes of nobelium and isotopes of lawrencium (and of heavier elements) have relatively short half-lives. For nobelium 11 isotopes are known with mass numbers 250–260 and 262. The chemical properties of nobelium and lawrencium were studied with 255No (t1/2 = 3 min) and 256Lr (t1/2 = 35 s). The longest-lived nobelium isotope 259No has a half-life of 1.5 hours.[56]
Distribution in nature[edit]

Unprocessed uranium ore
Thorium and uranium are the most abundant actinides in nature with the respective mass concentrations of 1.6×10−3% and 4×10−4%.[67]Uranium mostly occurs in the Earth’s crust as a mixture of its oxides in the minerals uraninite, which is also called pitchblende because of its black color. There are several dozens of other uranium minerals such as carnotite (KUO2VO4·3H2O) and autunite(Ca(UO2)2(PO4)2·nH2O). The isotopic composition of natural uranium is 238U (relative abundance 99.2742%), 235U (0.7204%) and 234U(0.0054%); of these 238U has the largest half-life of 4.51×109 years.[68][69] The worldwide production of uranium in 2009 amounted to 50,572 tonnes, of which 27.3% was mined in Kazakhstan. Other important uranium mining countries are Canada (20.1%), Australia (15.7%), Namibia (9.1%), Russia (7.0%), and Niger (6.4%).[70]
| Ore | Location | Uranium content, % |
Mass ratio 239Pu/ore |
Ratio 239Pu/U (×1012) |
|---|---|---|---|---|
| Uraninite | Canada | 13.5 | 9.1×10−12 | 7.1 |
| Uraninite | Congo | 38 | 4.8×10−12 | 12 |
| Uraninite | Colorado, US | 50 | 3.8×10−12 | 7.7 |
| Monazite | Brazil | 0.24 | 2.1×10−14 | 8.3 |
| Monazite | North Carolina, US | 1.64 | 5.9×10−14 | 3.6 |
| Fergusonite | – | 0.25 | <1×10−14 | <4 |
| Carnotite | – | 10 | <4×10−14 | <0.4 |
The most abundant thorium minerals are thorianite(ThO2), thorite (ThSiO4) and monazite, ((Th,Ca,Ce)PO4). Most thorium minerals contain uranium and vice versa; and they all have significant fraction of lanthanides. Rich deposits of thorium minerals are located in the United States (440,000 tonnes), Australia and India (~300,000 tonnes each) and Canada (~100,000 tonnes).[72]
The abundance of actinium in the Earth’s crust is only about 5×10−15%.[61] Actinium is mostly present in uranium-containing, but also in other minerals, though in much smaller quantities. The content of actinium in most natural objects corresponds to the isotopic equilibrium of parent isotope 235U, and it is not affected by the weak Ac migration.[29] Protactinium is more abundant (10−12%) in the Earth’s crust than actinium. It was discovered in the uranium ore in 1913 by Fajans and Göhring.[33] As actinium, the distribution of protactinium follows that of 235U.[61]
The half-life of the longest-lived isotope of neptunium, 237Np, is negligible compared to the age of the Earth. Thus neptunium is present in nature in negligible amounts produced as intermediate decay products of other isotopes.[37] Traces of plutonium in uranium minerals were first found in 1942, and the more systematic results on 239Pu are summarized in the table (no other plutonium isotopes could be detected in those samples). The upper limit of abundance of the longest-living isotope of plutonium, 244Pu, is 3×10−20%. Plutonium could not be detected in samples of lunar soil. Owing to its scarcity in nature, most plutonium is produced synthetically.[71]
Extraction[edit]

Monazite—a major thorium mineral
Owing to the low abundance of actinides, their extraction is a complex, multistep process. Fluorides of actinides are usually used because they are insoluble in water and can be easily separated with redox reactions. Fluorides are reduced with calcium, magnesium orbarium:[73]


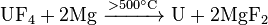
Among the actinides, thorium and uranium are the easiest to isolate. Thorium is extracted mostly from monazite: thorium diphosphate (Th(PO4)2) is reacted with nitric acid, and the produced thorium nitrate treated with tributyl phosphate. Rare-earth impurities are separated by increasing the pH in sulfate solution.[73]
In another extraction method, monazite is decomposed with a 45% aqueous solution of sodium hydroxide at 140 °C. Mixed metal hydroxides are extracted first, filtered at 80 °C, washed with water and dissolved with concentrated hydrochloric acid. Next, the acidic solution is neutralized with hydroxides to pH = 5.8 that results in precipitation of thorium hydroxide (Th(OH)4) contaminated with ~3% of rare-earth hydroxides; the rest of rare-earth hydroxides remains in solution. Thorium hydroxide is dissolved in an inorganic acid and then purified from the rare earth elements. An efficient method is the dissolution of thorium hydroxide in nitric acid, because the resulting solution can be purified byextraction with organic solvents:[73]
Separation of uranium and plutonium from nuclear fuel[74]
Th(OH)4 + 4 HNO3 → Th(NO3)4 + 4 H2O
Metallic thorium is separated from the anhydrous oxide, chloride or fluoride by reacting it with calcium in an inert atmosphere:[75]
ThO2 + 2 Ca → 2 CaO + Th
Sometimes thorium is extracted by electrolysis of a fluoride in a mixture of sodium and potassium chloride at 700–800 °C in a graphitecrucible. Highly pure thorium can be extracted from its iodide with the crystal bar process.[76]
Uranium is extracted from its ores in various ways. In one method, the ore is burned and then reacted with nitric acid to convert uranium into a dissolved state. Treating the solution with a solution of tributyl phosphate (TBP) in kerosene transforms uranium into an organic form UO2(NO3)2(TBP)2. The insoluble impurities are filtered and the uranium is extracted by reaction with hydroxides as (NH4)2U2O7 or with hydrogen peroxide as UO4·2H2O.[73]
When the uranium ore is rich in such minerals as dolomite, magnesite, etc., those minerals consume much acid. In this case, the carbonate method is used for uranium extraction. Its main component is an aqueous solution of sodium carbonate, which converts uranium into a complex [UO2(CO3)3]4−, which is stable in aqueous solutions at low concentrations of hydroxide ions. The advantages of the sodium carbonate method are that the chemicals have low corrosivity (compared to nitrates) and that most non-uranium metals precipitate from the solution. The disadvantage is that tetravalent uranium compounds precipitate as well. Therefore, the uranium ore is treated with sodium carbonate at elevated temperature and under oxygen pressure:
- 2 UO2 + O2 + 6 CO2−
3 → 2 [UO2(CO3)3]4−
This equation suggests that the best solvent for the uranium carbonate processing is a mixture of carbonate with bicarbonate. At high pH, this results in precipitation of diuranate, which is treated with hydrogen in the presence of nickel yielding an insoluble uranium tetracarbonate.[73]
Another separation method uses polymeric resins as a polyelectrolyte. Ion exchange processes in the resins result in separation of uranium. Uranium from resins is washed with a solution of ammonium nitrate or nitric acid that yields uranyl nitrate, UO2(NO3)2·6H2O. When heated, it turns into UO3, which is converted to UO2 with hydrogen:
- UO3 + H2 → UO2 + H2O
Reacting uranium dioxide with fluoric acid changes it to uranium tetrafluoride, which yields uranium metal upon reaction with magnesium metal:[75]
- 4 HF + UO2 → UF4 + 2 H2O
To extract plutonium, neutron-irradiated uranium is dissolved in nitric acid, and a reducing agent (FeSO4, or H2O2) is added to the resulting solution. This addition changes the oxidation state of plutonium from +6 to +4, while uranium remains in the form of uranyl nitrate (UO2(NO3)2). The solution is treated with a reducing agent and neutralized withammonium carbonate to pH = 8 that results in precipitation of Pu4+ compounds.[73]
In another method, Pu4+ and UO2+
2 are first extracted with tributyl phosphate, then reacted with hydrazine washing out the recovered plutonium.[73]
The major difficulty in separation of actinium is the similarity of its properties with those of lanthanum. Thus actinium is either synthesized in nuclear reactions from isotopes of radium or separated using ion-exchange procedures.[29]
Properties[edit]
Actinides have similar properties to lanthanides. The 6d and 7s electronic shells are filled in actinium and thorium, and the 5f shell is being filled with further increase in atomic number; the 4f shell is filled in the lanthanides. The first experimental evidence for the filling of the 5f shell in actinides was obtained by McMillan and Abelson in 1940.[77] As in lanthanides (see lanthanide contraction), the ionic radius of actinides monotonically decreases with atomic number (see also Aufbau principle).[78]
| [hide]Property | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Core charge | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 | 100 | 101 | 102 | 103 |
| atomic mass | [227] | 232.0377(4) | 231.03588(2) | 238.02891(3) | [237] | [244] | [243] | [247] | [247] | [251] | [252] | [257] | [258] | [259] | [266] |
| Number of natural isotopes[80] | 3 | 7 | 3 | 8 | 3 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Natural isotopes[80][81] | 225, 227–228 | 227–232, 234 | 231, 233–234 | 233–240 | 237, 239–240 | 238–240, 244 | — | — | — | — | — | — | — | — | — |
| Longest-lived isotope | 227 | 232 | 231 | 238 | 237 | 244 | 243 | 247 | 247 | 251 | 252 | 257 | 258 | 259 | 266 |
| Half-life of the longest-lived isotope | 21.8 years | 14 billion years | 32,500 years | 4.47 billion years | 2.14 million years | 80.8 million years | 7,370 years | 15.6 million years | 1,400 years | 900 years | 1.29 years | 100.5 days | 52 days | 58 min | 11 hours |
| Electronic configuration in the ground state | 6d17s2 | 6d27s2 | 5f26d17s2or 5f16d27s2 | 5f36d17s2 | 5f46d17s2or 5f57s2 | 5f67s2 | 5f77s2 | 5f76d17s2 | 5f97s2or 5f86d17s2 | 5f107s2 | 5f117s2 | 5f127s2 | 5f137s2 | 5f147s2 | 5f147s27p1 |
| Oxidation states | 2, 3 | 2, 3, 4 | 2, 3, 4, 5 | 2, 3, 4, 5, 6 | 3, 4, 5, 6, 7 | 3, 4, 5, 6, 7, 8 | 2, 3, 4, 5, 6, 7 | 2, 3, 4, 6, 8 | 2, 3, 4 | 2, 3, 4 | 2, 3, 4 | 2, 3 | 2, 3 | 2, 3 | 3 |
| Metallic radius, nm | 0.203 | 0.180 | 0.162 | 0.153 | 0.150 | 0.162 | 0.173 | 0.174 | 0.170 | 0.186 | 0.186 | ? 0.198 | ? 0.194 | ? 0.197 | ? 0.171 |
| Ionic radius, nm: An4+ An3+ |
— 0.126 |
0.114 — |
0.104 0.118 |
0.103 0.118 |
0.101 0.116 |
0.100 0.115 |
0.099 0.114 |
0.099 0.112 |
0.097 0.110 |
0.096 0.109 |
0.085 0.098 |
0.084 0.091 |
0.084 0.090 |
0.084 0.095 |
0.083 0.088 |
| Temperature, °C: melting boiling |
1050 3198 |
1842 4788 |
1568 ? 4027 |
1132.2 4131 |
639 ? 4174 |
639.4 3228 |
1176 ? 2607 |
1340 3110 |
986 2627 |
900 ? 1470 |
860 ? 996 |
1530 — |
830 — |
830 — |
1630 — |
| Density, g/cm3 | 10.07 | 11.78 | 15.37 | 19.06 | 20.45 | 19.84 | 11.7 | 13.51 | 14.78 | 15.1 | 8.84 | ||||
| Standard electrode potential, V: E° (An4+/An0) E° (An3+/An0) |
— −2.13 |
−1.83 — |
−1.47 — |
−1.38 −1.66 |
−1.30 −1.79 |
−1.25 −2.00 |
−0.90 −2.07 |
−0.75 −2.06 |
−0.55 −1.96 |
−0.59 −1.97 |
−0.36 −1.98 |
−0.29 −1.96 |
— −1.74 |
— −1.20 |
— −2.10 |
| Color [M(H2O)n]4+ [M(H2O)n]3+ |
— Colorless |
Colorless Blue |
Yellow Dark blue |
Green Purple |
Yellow-green Purple |
Brown Violet |
Red Rose |
Yellow Colorless |
Beige Yellow-green |
Green Green |
— Pink |
— — |
— — |
— — |
— — |
| Oxidation state | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 |
| +3 | Ac3+ | Th3+ | Pa3+ | U3+ | Np3+ | Pu3+ | Am3+ | Cm3+ | Bk3+ | Cf3+ | Es3+ |
| +4 | Th4+ | Pa4+ | U4+ | Np4+ | Pu4+ | Am4+ | Cm4+ | Bk4+ | Cf4+ | ||
| +5 | PaO+ 2 |
UO+ 2 |
NpO+ 2 |
PuO+ 2 |
AmO+ 2 |
||||||
| +6 | UO2+ 2 |
NpO2+ 2 |
PuO2+ 2 |
AmO2+ 2 |
|||||||
| +7 | NpO3+ 2 |
PuO3+ 2 |
[AmO6]5− |
Physical properties[edit]
 |
 |
| Major crystal structures of some actinides vs. temperature | Metallic and ionic radii of actinides[79] |

A pellet of 238PuO2 to be used in aradioisotope thermoelectric generatorfor either the Cassini or Galileomission. The pellet produces 62 watts of heat and glows because of the heat generated by the radioactive decay (primarily α). Photo is taken after insulating the pellet under a graphiteblanket for minutes and removing the blanket.

Actinides are typical metals. All of them are soft and have a silvery color (but tarnish in air),[83] relatively high density and plasticity. Some of them can be cut with a knife. Their electrical resistivity varies between 15 and 150 µOhm·cm.[79] The hardness of thorium is similar to that of soft steel, so heated pure thorium can be rolled in sheets and pulled into wire. Thorium is nearly half as dense as uranium and plutonium, but is harder than either of them. All actinides are radioactive, paramagnetic, and, with the exception of actinium, have several crystalline phases: plutonium has seven, and uranium, neptunium and californium three. Crystal structures of protactinium, uranium, neptunium and plutonium do not have clear analogs among the lanthanides and are more similar to those of the 3d–transition metals.[69]
All actinides are pyrophoric, especially when finely divided, that is, they spontaneously ignite upon reaction with air.[83] The melting point of actinides does not have a clear dependence on the number of f-electrons. The unusually low melting point of neptunium and plutonium (~640 °C) is explained by hybridization of 5f and 6d orbitals and the formation of directional bonds in these metals.[69]
| [show]Lanthanides | Ln3+, Å | Actinides | An3+, Å | An4+, Å |
|---|
Chemical properties[edit]
Like the lanthanides, all actinides are highly reactive with halogens and chalcogens; however, the actinides react more easily. Actinides, especially those with a small number of 5f-electrons, are prone to hybridization. This is explained by the similarity of the electron energies at the 5f, 7s and 6d shells. Most actinides exhibit a larger variety of valence states, and the most stable are +6 for uranium, +5 for protactinium and neptunium, +4 for thorium and plutonium and +3 for actinium and other actinides.[85]
Chemically, actinium is similar to lanthanum, which is explained by their similar ionic radii and electronic structure. Like lanthanum, actinium has oxidation of +3, but it is less reactive and has more pronounced basic properties. Among other trivalent actinides Ac3+ is least acidic, i.e. has the weakest tendency to hydrolyze in aqueous solutions.[29][69]
Thorium is rather active chemically. Owing to lack of electrons on 6d and 5f orbitals, the tetravalent thorium compounds are colorless. At pH < 3, the solutions of thorium salts are dominated by the cations [Th(H2O)8]4+. The Th4+ ion is relatively large, and depending on the coordination number can have a radius between 0.95 and 1.14 Å. As a result, thorium salts have a weak tendency to hydrolyse. The distinctive ability of thorium salts is their high solubility, not only in water, but also in polar organic solvents.[69]
Protactinium exhibits two valence states; the +5 is stable, and the +4 state easily oxidizes to protactinium(V). Thus tetravalent protactinium in solutions is obtained by the action of strong reducing agents in a hydrogen atmosphere. Tetravalent protactinium is chemically similar to uranium(IV) and thorium(IV). Fluorides, phosphates, hypophosphate, iodate and phenylarsonates of protactinium(IV) are insoluble in water and dilute acids. Protactinium forms soluble carbonates. The hydrolytic properties of pentavalent protactinium are close to those of tantalum(V) and niobium(V). The complex chemical behavior of protactinium is a consequence of the start of the filling of the 5f shell in this element.[61]
Uranium has a valence from 3 to 6, the last being most stable. In the hexavalent state, uranium is very similar to the group 6 elements. Many compounds of uranium(IV) and uranium(VI) are non-stoichiometric, i.e. have variable composition. For example, the actual chemical formula of uranium dioxide is UO2+x, where x varies between −0.4 and 0.32. Uranium(VI) compounds are weak oxidants. Most of them contain the linear “uranyl” group, UO2+
2. Between 4 to 6 ligands can be accommodated in an equatorial plane perpendicular to the uranyl group. The uranyl group acts as a hard acid and forms stronger complexes with oxygen-donor ligands than with nitrogen-donor ligands. NpO2+
2 andPuO2+
2 are also the common form of Np and Pu in the +6 oxidation state. Uranium(IV) compounds exhibit reducing properties, e.g., they are easily oxidized by atmospheric oxygen. Uranium(III) is a very strong reducing agent. Owing to the presence of d-shell, uranium (as well as many other actinides) forms organometallic compounds, such as UIII(C5H5)3 and UIV(C5H5)4.[69][86]
Neptunium has valence states from 3 to 7, which can be simultaneously observed in solutions. The most stable state in solution is +5, but the valence +4 is preferred in solid neptunium compounds. Neptunium metal is very reactive. Ions of neptunium are prone to hydrolysis and formation of coordination compounds.[37]
Plutonium also exhibits valence states between 3 and 7 inclusive, and thus is chemically similar to neptunium and uranium. It is highly reactive, and quickly forms an oxide film in air. Plutonium reacts with hydrogen even at temperatures as low as 25–50 °C; it also easily forms halides and intermetallic compounds. Hydrolysis reactions of plutonium ions of different oxidation states are quite diverse. Plutonium(V) can enter polymerization reactions.[87][88]
The largest chemical diversity among actinides is observed in americium, which can have valence between 2 and 6. Divalent americium is obtained only in dry compounds and non-aqueous solutions (acetonitrile). Oxidation states +3, +5 and +6 are typical for aqueous solutions, but also in the solid state. Tetravalent americium forms stable solid compounds (dioxide, fluoride and hydroxide) as well as complexes in aqueous solutions. It was reported that in alkaline solution americium can be oxidized to the heptavalent state, but these data proved erroneous. The most stable valence of americium is 3 in the aqueous solutions and 3 or 4 in solid compounds.[89]
Valence 3 is dominant in all subsequent elements up to lawrencium (with the possible exception of nobelium). Curium can be tetravalent in solids (fluoride, dioxide). Berkelium, along with a valence of +3, also shows the valence of +4, more stable than that of curium; the valence 4 is observed in solid fluoride and dioxide. The stability of Bk4+ in aqueous solution is close to that of Ce4+. Only valence 3 was observed for californium, einsteinium and fermium. The divalent state is proven for mendelevium and nobelium, and in nobelium it is more stable than the trivalent state. Lawrencium shows valence 3 both in solutions and solids.[89]
The redox potential  increases from −0.32 V in uranium, through 0.34 V (Np) and 1.04 V (Pu) to 1.34 V in americium revealing the increasing reduction ability of the An4+ion from americium to uranium. All actinides form AnH3 hydrides of black color with salt-like properties. Actinides also produce carbides with the general formula of AnC or AnC2(U2C3 for uranium) as well as sulfides An2S3 and AnS2.[85]
increases from −0.32 V in uranium, through 0.34 V (Np) and 1.04 V (Pu) to 1.34 V in americium revealing the increasing reduction ability of the An4+ion from americium to uranium. All actinides form AnH3 hydrides of black color with salt-like properties. Actinides also produce carbides with the general formula of AnC or AnC2(U2C3 for uranium) as well as sulfides An2S3 and AnS2.[85]
-

Uranyl nitrate (UO2(NO3)2)
-

Aqueous solutions of uranium III, IV, V, VI salts
-

Aqueous solutions of neptunium III, IV, V, VI, VII salts
-

Aqueous solutions of plutonium III, IV, V, VI, VII salts
-

U3O8 (yellowcake)







Compounds[edit]
Oxides and hydroxides[edit]
| [show]Compound | Color | Crystal symmetry, type | Lattice constants, Å | Density, g/cm3 | Temperature, °C | ||
|---|---|---|---|---|---|---|---|
| [show]Oxidation state | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 |
|---|
| Chemical formula | ThO2 | PaO2 | UO2 | NpO2 | PuO2 | AmO2 | CmO2 | BkO2 | CfO2 |
| CAS-number | 1314-20-1 | 12036-03-2 | 1344-57-6 | 12035-79-9 | 12059-95-9 | 12005-67-3 | 12016-67-0 | 12010-84-3 | 12015-10-0 |
| Molar mass | 264.04 | 263.035 | 270.03 | 269.047 | 276.063 | 275.06 | 270–284** | 279.069 | 283.078 |
| Melting point[95] | 3390 °C | 2865 °C | 2547 °C | 2400 °C | 2175 °C | ||||
| Crystal structure |  An4+: __ / O2−: __ |
||||||||
| Space group | Fm3m | ||||||||
| Coordination number | An[8], O[4] | ||||||||
- An – actinide
**Depending on the isotopes
Some actinides can exists in several oxide forms such as An2O3, AnO2, An2O5 and AnO3. For all actinides, oxides AnO3 are amphoteric and An2O3, AnO2 and An2O5 are basic, they easily react with water, forming bases:[85]
- An2O3 + 3 H2O → 2 An(OH)3.
These bases are poorly soluble in water and by their activity are close to the hydroxides of rare-earth metals. The strongest base is of actinium. All compounds of actinium are colorless, except for black actinium sulfide (Ac2S3).[85] Dioxides of tetravalent actinides crystallize in the cubic system, same as in calcium fluoride.
Thorium reacting with oxygen exclusively forms dioxide:

Thorium dioxide is a refractory material with the highest melting point among any known oxide (3390 °C).[94] Adding 0.8–1% ThO2 to tungsten stabilizes its structure, so the doped filaments have better mechanical stability to vibrations. To dissolve ThO2 in acids, it is heated to 500–600 °C; heating above 600 °C produces a very resistant to acids and other reagents form of ThO2. Small addition of fluoride ions catalyses dissolution of thorium dioxide in acids.
Two protactinium oxides were obtained: PaO2 (black) and Pa2O5(white); the former is isomorphic with ThO2 and the latter is easier to obtain. Both oxides are basic, and Pa(OH)5is a weak, poorly soluble base.[85]
Decomposition of certain salts of uranium, for example UO2(NO3)·6H2O in air at 400 °C, yields orange or yellow UO3.[94] This oxide is amphoteric and forms several hydroxides, the most stable being UO2(OH)2. Reaction of uranium(VI) oxide with hydrogen results in uranium dioxide, which is similar in its properties with ThO2. This oxide is also basic and corresponds to the uranium hydroxide (U(OH)4).[85]
Plutonium, neptunium and americium form two basic oxides: An2O3 and AnO2. Neptunium trioxide is unstable; thus, only Np3O8 could be obtained so far. However, the oxides of plutonium and neptunium with the chemical formula AnO2 and An2O3 are well characterized.[85]
Salts[edit]
| Chemical formula | AcCl3 | UCl3 | NpCl3 | PuCl3 | AmCl3 | CmCl3 | BkCl3 | CfCl3 |
| CAS-number | 22986-54-5 | 10025-93-1 | 20737-06-8 | 13569-62-5 | 13464-46-5 | 13537-20-7 | 13536-46-4 | 13536-90-8 |
| Molar mass | 333.386 | 344.387 | 343.406 | 350.32 | 349.42 | 344–358** | 353.428 | 357.438 |
| Melting point | 837 °C | 800 °C | 767 °C | 715 °C | 695 °C | 603 °C | 545 °C | |
| Boiling point | 1657 °C | 1767 °C | 850 °C | |||||
| Crystal structure |  An3+: __ / Cl−: __ |
|||||||
| Space group | P63/m | |||||||
| Coordination number | An*[9], Cl [3] | |||||||
| Lattice constants | a = 762 pm c = 455 pm |
a = 745.2 pm c = 432.8 pm |
a = 739.4 pm c = 424.3 pm |
a = 738.2 pm c = 421.4 pm |
a = 726 pm c = 414 pm |
a = 738.2 pm c = 412.7 pm |
a = 738 pm c = 409 pm |
|
- *An – actinide
**Depending on the isotopes
| [show]Compound | Color | Crystal symmetry, type | Lattice constants, Å | Density, g/cm3 | ||
|---|---|---|---|---|---|---|
Einsteinium triiodide glowing in the dark
Actinides easily react with halogens forming salts with the formulas MX3 and MX4 (X = halogen). So the first berkelium compound, BkCl3, was synthesized in 1962 with an amount of 3 nanograms. Like the halogens of rare earth elements, actinide chlorides, bromides, andiodides are water-soluble, and fluorides are insoluble. Uranium easily yields a colorless hexafluoride, which sublimates at a temperature of 56.5 °C; because of its volatility, it is used in the separation of uranium isotopes with gas centrifuge or gaseous diffusion. Actinide hexafluorides have properties close to anhydrides. They are very sensitive to moisture and hydrolyze forming AnO2F2.[99] The pentachloride and black hexachloride of uranium were synthesized, but they are both unstable.[85]
Action of acids on actinides yields salts, and if the acids are non-oxidizing then the actinide in the salt is in low-valence state:
- U + 2 H2SO4 → U (SO4)2 + 2 H2
- 2 Pu + 6 HCl → 2 PuCl3 + 3 H2
However, in these reactions the regenerating hydrogen can react with the metal, forming the corresponding hydride. Uranium reacts with acids and water much more easily than thorium.[85]
Actinide salts can also be obtained by dissolving the corresponding hydroxides in acids. Nitrates, chlorides, sulfates and perchlorates of actinides are water-soluble. When crystallizing from aqueous solutions, these salts forming a hydrates, such as Th(NO3)4·6H2O, Th(SO4)2·9H2O and Pu2(SO4)3·7H2O. Salts of high-valence actinides easily hydrolyze. So, colorless sulfate, chloride, perchlorate and nitrate of thorium transform into basic salts with formulas Th(OH)2SO4 and Th(OH)3NO3. The solubility and insolubility of trivalent and tetravalent actinides is like that of lanthanide salts. So phosphates, fluorides, oxalates, iodates and carbonates of actinides are weakly soluble in water; they precipitate as hydrates, such as ThF4·3H2O and Th(CrO4)2·3H2O.[85]
Actinides with oxidation state +6, except for the AnO22+-type cations, form [AnO4]2−, [An2O7]2− and other complex anions. For example, uranium, neptunium and plutonium form salts of the Na2UO4 (uranate) and (NH4)2U2O7 (diuranate) types. In comparison with lanthanides, actinides more easily form coordination compounds, and this ability increases with the actinide valence. Trivalent actinides do not form fluoride coordination compounds, whereas tetravalent thorium forms K2ThF6, KThF5, and even K5ThF9 complexes. Thorium also forms the corresponding sulfates (for example Na2SO4·Th (SO4)2·5H2O), nitrates and thiocyanates. Salts with the general formula An2Th(NO3)6·nH2O are of coordination nature, with the coordination number of thorium equal to 12. Even easier is to produce complex salts of pentavalent and hexavalent actinides. The most stable coordination compounds of actinides – tetravalent thorium and uranium – are obtained in reactions with diketones, e.g. acetylacetone.[85]
Applications[edit]

Interior of a smoke detectorcontaining americium-241.
While actinides have some established daily-life applications, such as in smoke detectors (americium)[100][101] and gas mantles(thorium),[75] they are mostly used in nuclear weapons and use as a fuel in nuclear reactors.[75] The last two areas exploit the property of actinides to release enormous energy in nuclear reactions, which under certain conditions may become self-sustaining chain reaction.

Self-illumination of a nuclear reactor by Cherenkov radiation.
The most important isotope for nuclear power applications is uranium-235. It is used in the thermal reactor, and its concentration in natural uranium does not exceed 0.72%. This isotope strongly absorbsthermal neutrons releasing much energy. One fission act of 1 gram of 235U converts into about 1 MW·day. Of importance, is that 235U emits more neutrons than it absorbs;[102] upon reaching the critical mass, 235U enters into a self-sustaining chain reaction.[69] Typically, uranium nucleus is divided into two fragments with the release of 2–3 neutrons, for example:
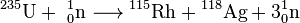
Other promising actinide isotopes for nuclear power are thorium-232 and its product from the thorium fuel cycle, uranium-233.
| Nuclear reactor[69][103][104] |
The core of most Generation II nuclear reactors contains a set of hollow metal rods, usually made of zirconium alloys, filled with solid nuclear fuel pellets – mostly oxide, carbide, nitride or monosulfide of uranium, plutonium or thorium, or their mixture (the so-called MOX fuel). The most common fuel is oxide of uranium-235.
 Fast neutrons are slowed by moderators, which contain water, carbon, deuterium, or beryllium, as thermal neutrons to increase the efficiency of their interaction with uranium-235. The rate of nuclear reaction is controlled by introducing additional rods made of boronor cadmium or a liquid absorbent, usually boric acid. Reactors for plutonium production are called breeder reactor or breeders; they have a different design and use fast neutrons. |
Emission of neutrons during the fission of uranium is important not only for maintaining the nuclear chain reaction, but also for the synthesis of the heavier actinides. Uranium-239 converts via β-decay into plutonium-239, which, like uranium-235, is capable of spontaneous fission. The world’s first nuclear reactors were built not for energy, but for producing plutonium-239 for nuclear weapons.
About half of the produced thorium is used as the light-emitting material of gas mantles.[75] Thorium is also added into multicomponent alloys of magnesium and zinc. So the Mg-Th alloys are light and strong, but also have high melting point and ductility and thus are widely used in the aviation industry and in the production of missiles. Thorium also has good electron emission properties, with long lifetime and low potential barrier for the emission.[102] The relative content of thorium and uranium isotopes is widely used to estimate the age of various objects, including stars (see radiometric dating).[105]
The major application of plutonium has been in nuclear weapons, where the isotope plutonium-239 was a key component due to its ease of fission and availability. Plutonium-based designs allow reducing thecritical mass to about a third of that for uranium-235.[106] The “Fat Man“-type plutonium bombs produced during the Manhattan Project used explosive compression of plutonium to obtain significantly higher densities than normal, combined with a central neutron source to begin the reaction and increase efficiency. Thus only 6.2 kg of plutonium was needed for an explosive yieldequivalent to 20 kilotons of TNT.[107] (See also Nuclear weapon design.) Hypothetically, as little as 4 kg of plutonium—and maybe even less—could be used to make a single atomic bomb using very sophisticated assembly designs.[108]
Plutonium-238 is potentially more efficient isotope for nuclear reactors, since it has smaller critical mass than uranium-235, but it continues to release much thermal energy (0.56 W/g)[101][109] by decay even when the fission chain reaction is stopped by control rods. Its application is limited by the high price (about 1000 USD/g). This isotope has been used in thermopiles and water distillation systems of some space satellites and stations. So Galileo and Apollo spacecraft (e.g. Apollo 14[110]) had heaters powered by kilogram quantities of plutonium-238 oxide; this heat is also transformed into electricity with thermopiles. The decay of plutonium-238 produces relatively harmless alpha particles and is not accompanied by gamma-irradiation. Therefore, this isotope (~160 mg) is used as the energy source in heart pacemakers where it lasts about 5 times longer than conventional batteries.[101]
Actinium-227 is used as a neutron source. Its high specific energy (14.5 W/g) and the possibility of obtaining significant quantities of thermally stable compounds are attractive for use in long-lasting thermoelectric generators for remote use. 228Ac is used as an indicator of radioactivity in chemical research, as it emits high-energy electrons (2.18 MeV) that can be easily detected. 228Ac–228Ra mixtures are widely used as an intense gamma-source in industry and medicine.[29]
Development of self-glowing actinide-doped materials with durable crystalline matrices is a new area of actinide utilization as the addition of alpha-emitting radionuclides to some glasses and crystals may confer luminescence.[111]
Toxicity[edit]

Schematic illustration of penetration of radiation through sheets of paper, aluminium and lead brick

Periodic table with elements colored according to the half-life of their most stable isotope.
Elements which contain at least one stable isotope.
Slightly radioactive elements: the most stable isotope is very long-lived, with a half-life of over four million years.
Moderately radioactive elements: the most stable isotope has half-life between 800 and 34,000 years.
Very radioactive elements: the most stable isotope has half-life between one day and 103 years.
Highly radioactive elements: the most stable isotope has half-life between several minutes and one day.
Extremely radioactive elements: the most stable isotope has half-life less than several minutes. Very little is known about these elements due to their extreme instability and radioactivity.
Radioactive substances can harm human health via (i) local skin contamination, (ii) internal exposure due to ingestion of radioactive isotopes, and (iii) external overexposure by β-activity and γ-radiation. Together with radium and transuranium elements, actinium is one of the most dangerous radioactive poisons with high specific α-activity. The most important feature of actinium is its ability to accumulate and remain in the surface layer of skeletons. At the initial stage of poisoning, actinium accumulates in the liver. Another danger of actinium is that it undergoes radioactive decay faster than being excreted. Adsorption from the digestive tract is much smaller (~0.05%) for actinium than radium.[29]
Protactinium in the body tends to accumulate in the kidneys and bones. The maximum safe dose of protactinium in the human body is 0.03 µCi that corresponds to 0.5 micrograms of 231Pa. This isotope, which might be present in the air as aerosol, is 2.5×108 times more toxic than hydrocyanic acid.[61]
Plutonium, when entering the body through air, food or blood (e.g. a wound), mostly settles in the lungs, liver and bones with only about 10% going to other organs, and remains there for decades. The long residence time of plutonium in the body is partly explained by its poor solubility in water. Some isotopes of plutonium emit ionizing α-radiation, which damages the surrounding cells. Themedian lethal dose (LD50) for 30 days in dogs after intravenous injection of plutonium is 0.32 milligram per kg of body mass, and thus the lethal dose for humans is approximately 22 mg for a person weighing 70 kg; the amount for respiratory exposure should be approximately four times greater. Another estimate assumes that plutonium is 50 times less toxic than radium, and thus permissible content of plutonium in the body should be 5 µg or 0.3 µCi. Such amount is nearly invisible in under microscope. After trials on animals, this maximum permissible dose was reduced to 0.65 µg or 0.04 µCi. Studies on animals also revealed that the most dangerous plutonium exposure route is through inhalation, after which 5–25% of inhaled substances is retained in the body. Depending on the particle size and solubility of the plutonium compounds, plutonium is localized either in the lungs or in the lymphatic system, or is absorbed in the blood and then transported to the liver and bones. Contamination via food is the least likely way. In this case, only about 0.05% of soluble 0.01% insoluble compounds of plutonium absorbs into blood, and the rest is excreted. Exposure of damaged skin to plutonium would retain nearly 100% of it.[87]
Using actinides in nuclear fuel, sealed radioactive sources or advanced materials such as self-glowing crystals has many potential benefits. However, a serious concern is the extremely high radiotoxicity of actinides and their migration in the environment.[112] Use of chemically unstable forms of actinides in MOX and sealed radioactive sources is not appropriate by modern safety standards. There is a challenge to develop stable and durable actinide-bearing materials, which provide safe storage, use and final disposal. A key need is application of actinide solid solutions in durable crystalline host phases.[111]
Post-transition metal
From Wikipedia, the free encyclopedia
Post-transition metals in the periodic table
Elements classified as post-transition metals by Masterton, Hurley and Neth:[1] Ga, In, Tl, Sn, Pb, Bi, Po
Also recognised by Deming:[4] Cu, Ag, Au (but he counted Al and groups 1 and 2 as ‘light metals’)[n 1]
Elements that might be post-transition metals: At, Cn, Uut, Fl, Uup, Lv, Uus
In chemistry, post-transition metals are the metallic elements in the periodic table located between the transition metals (to their left) and the metalloids (to their right). Usually included in this category are gallium, indium and thallium;tin and lead; and bismuth. Which elements are counted as post-transition metals depends, in periodic table terms, on where the transition metals are taken to end and where the metalloids or non-metals are taken to start.
Physically, post-transition metals are soft (or brittle), have poor mechanical strength, and melting points lower than those of the transition metals; most also have boiling points lower than those of the transition metals. Being close to the metal-nonmetal border, their crystalline structures tend to showcovalent or directional bonding effects, having generally greater complexity or fewer nearest neighbours than other metallic elements.
Chemically, they are characterised—to varying degrees—by covalent bonding tendencies, acid-base amphoterism and the formation of anionic species such as aluminates, stannates, and bismuthates (in the case of aluminium, tin, and bismuth, respectively). They can also form Zintl phases (half-metallic compounds formed between highly electropositive metals and moderatelyelectronegative metals or metalloids).
The expression ‘post-transition metals’ is used here as there is no IUPAC-approved collective name for these metals. Occasionally, some or all of them have instead been referred to as B-subgroup metals, other metals, or p-block metals; and by at least eleven other alternative labels. All these labels are surveyed later in this article.
Applicable elements[edit]

Scatter plot of electronegativity values and melting points for metals (up to fermium, element 100) and some borderline elements (Ge, As, Sb, At). Elements categorised by some authors as post-transition metals are distinguished by their relatively high electronegativity values and relatively low melting points. High electronegativity corresponds to increasing nonmetallic character;[8] low melting temperature corresponds to weaker cohesive forces between atoms and reduced mechanical strength.[9] The geography of the plot broadly matches that of the periodic table. Starting from the bottom left, and proceeding clockwise, the alkali metals are followed by the heavier alkaline earth metals; the rare earths and actinides (Sc, Y and the lanthanides being here treated as rare earths); transition metals with intermediate electronegativity values and melting points; therefractory metals; the platinum group metals; and the coinage metals leading and forming a part of the post-transition metals.
The increased electropositivity of Be and Mg and the higher melting point of Be distances these light alkaline earth metals from their heavier congeners. This separation extends to other differences in physical and chemical behaviour between the light and heavier alkaline earth metals.[n 2]
Usually included in this category are the group 13–15 metals: gallium, indium and thallium; tin and lead; and bismuth. Other elements sometimes included are copper, silver and gold (which are usually considered to be transition metals); zinc, cadmium and mercury (which are otherwise considered to be transition metals); and aluminium, germanium, arsenic and antimony (the latter three of which are usually considered to be metalloids). Astatine, which is usually classified as a nonmetal or a metalloid, has been predicted to have a metallic crystalline structure. If so, it would be a post-transition metal. Elements 113–117 may be post-transition metals; insufficient quantities of them have been synthesized to allow investigation of their actual physical and chemical properties.
Which elements start to be counted as post-transition metals depends, in periodic table terms, on where the transition metals are taken to end.[n 3] In the 1950s, most inorganic chemistry textbooks defined transition elements as finishing at group 10 (nickel, palladium andplatinum), therefore excluding group 11 (copper, silver and gold), and group 12 (zinc,cadmium and mercury). A survey of chemistry books in 2003 showed that the transition metals ended at either group 11 or group 12 with roughly equal frequency.[13] Where the post-transition metals end depends on where the metalloids or nonmetals start. Boron, silicon, germanium, arsenic, antimony and tellurium are commonly recognised as metalloids; other authors treat some or all of these elements as nonmetals.
Rationale[edit]
The diminished metallic nature of the post-transition metals is largely attributable to the increase in nuclear charge going across the periodic table, from left to right.[14] The increase in nuclear charge is partially offset by an increasing number of electrons but as these are spatially distributed each extra electron does not fully screen each successive increase in nuclear charge, and the latter therefore dominates.[15] With some irregularities, atomic radii contract, ionisation energies increase,[14] fewer electrons become available for metallic bonding,[16] and “ions [become] smaller and more polarizing and more prone to covalency.”[17] This phenomenon is more evident in period 4–6 post-transition metals, due to inefficient screening of their nuclear charges by their d10 and (in the case of the period 6 metals) f14 electron configurations;[18] the screening power of electrons decreases in the sequence s > p > d > f. The reductions in atomic size due to the interjection of the d- and f-blocks are referred to as, respectively, the ‘scandide’ or ‘d-block contraction‘,[n 4] and the ‘lanthanide contraction‘.[19] Relativistic effects also “increase the binding energy”, and hence ionisation energy, of the electrons in “the 6s shell in gold and mercury, and the 6p shell in subsequent elements of period 6.”[20]
Nomenclature[edit]
Origin[edit]
The origin of the term post-transition metal is unclear. An early usage is recorded by Deming, in 1940, in his well-known[21] book Fundamental Chemistry.[4] He treated the transition metals as finishing at group 10 (nickel, palladium and platinum). He referred to the ensuing elements in periods 4 to 6 of the periodic table (copper to germanium; silver to antimony; gold to polonium)—in view of their underlying d10 electronic configurations—as post-transition metals.
Descriptive chemistry[edit]
- This section outlines relevant physical and chemical properties of the elements typically or sometimes classified as post-transition metals. For complete profiles, including history, production, specific uses, and biological roles and precautions, see the main article for each element. Abbreviations: MH—Mohs hardness; BCN—bulk coordination number.[n 5]
Group 11[edit]
Main article: Group 11 element
The group 11 metals are typically categorised as transition metals given they can form ions with incomplete d-shells. Physically, they have the relatively low melting points and high electronegativity values associated with post-transition metals. “The filled d subshell and free s electron of Cu, Ag, and Au contribute to their high electrical and thermal conductivity. Transition metals to the left of group 11 experience interactions between s electrons and the partially filled d subshell that lower electron mobility.”[24] Chemically, the group 11 metals in their +1 valence states show similarities to other post-transition metals;[25] they are occasionally classified as such.[26]

Copper

Silver

Gold
Copper is a soft metal (MH 2.5–3.0)[27] with low mechanical strength.[28] It has a close-packed face-centred cubic structure (BCN 12).[29] Copper behaves like a transition metal in its preferred oxidation state of +2. Stable compounds in which copper is in its less preferred oxidation state of +1 (Cu2O, CuCl, CuBr, CuI and CuCN, for example) have significant covalent character.[30] The oxide (CuO) is amphoteric, with predominating basic properties; it can be fused with alkali oxides (M2O; M = Na, K) to give anionic oxycuprates (M2CuO2).[31] Copper forms Zintl phases such as Li7CuSi2[32] and M3Cu3Sb4 (M = Y, La, Ce, Pr, Nd, Sm, Gd, Tb, Dy, Ho, or Er).[33]
Silver is a soft metal (MH 2.5–3)[34] with low mechanical strength.[35] It has a close-packed face-centred cubic structure (BCN 12).[36] The chemistry of silver is dominated by its +1 valence state in which it shows generally similar physical and chemical properties to compounds of thallium, a main group metal, in the same oxidation state.[37] It tends to bond covalently in most of its compounds.[38] The oxide (Ag2O) is amphoteric, with basic properties predominating.[39] Silver forms a series of oxoargentates (M3AgO2, M = Na, K, Rb).[40] It is a constituent of Zintl phases such as Li2AgM (M = Al, Ga, In, Tl, Si, Ge, Sn or Pb)[41]and Yb3Ag2.[42]
Gold is a soft metal (MH 2.5–3)[43] that is easily deformed.[44] It has a close-packed face-centred cubic structure (BCN 12).[36] The chemistry of gold is dominated by its +3 valence state; all such compounds of gold feature covalent bonding,[45] as do its stable +1 compounds.[46] Gold oxide (Au2O3) is amphoteric, with acid properties predominating; it forms anionic hydroxoaurates M[Au(OH)4] where M = Na, K, 1⁄2Ba, Tl; and aurates such as NaAuO2.[47] Gold is a constituent of Zintl phases such as M2AuBi (M = Li or Na);[48]Li2AuM (M = In, Tl, Ge, Pb, Sn)[49] and Ca5Au4.[42]
Group 12[edit]
Main article: Group 12 element
On the group 12 transition metals (zinc, cadmium and mercury), Smith[50] observed that, “Textbook writers have always found difficulty in dealing with these elements.” There is an abrupt and significant reduction in physical metallic character from group 11 to group 12.[51] Their chemistry is that of main group elements.[52] A 2003 survey of chemistry books showed that they were treated as either transition metals or main group elements on about a 50/50 basis.[13][n 6] The IUPAC Red Book notes that although the group 3−12 elements are commonly referred to as the transition elements, the group 12 elements are not always included.[54] The group 12 elements do not satisfy the IUPAC Gold Bookdefinition of a transition metal[55][n 7] (other than in the case of mercury at 4 K).

Zinc

Cadmium

Mercury
Zinc is a soft metal (MH 2.5) with poor mechanical properties.[57] It has a crystalline structure (BCN 6+6) that is slightly distorted from the ideal. Many zinc compounds are markedly covalent in character.[58] The oxides of zinc in its preferred oxidation state of +2, namely ZnO and Zn(OH)2, are amphoteric;[59] it forms anionic zincates in strongly basic solutions.[60] Zinc forms Zintl phases such as LiZn, NaZn13 and BaZn13.[61] Highly purified zinc, at room temperature, is ductile.[62] It reacts with moist air to form a thin layer of carbonate that prevents further corrosion.[63]
Cadmium is a soft, ductile metal (MH 2.0) that undergoes substantial deformation, under load, at room temperature.[64] Like zinc, it has a crystalline structure (BCN 6+6) that is slightly distorted from the ideal. The halides of cadmium, with the exception of the fluoride, exhibit a substantially covalent nature.[65] The oxides of cadmium in its preferred oxidation state of +2, namely CdO and Cd(OH)2, are weakly amphoteric; it forms cadmates in strongly basic solutions.[66] Cadmium forms Zintl phases such as LiCd, RbCd13 and CsCd13.[61] When heated in air to a few hundred degrees, cadmium represents a toxicity hazard due to the release of cadmium vapour; when heated to its boiling point in air (just above 1000 K; 725 C; 1340 F; cf steel ~2700 K; 2425 C; 4400 F),[67] the cadmium vapour oxidizes, ‘with a reddish-yellow flame, dispersing as an aerosol of potentially lethal CdO particles.’[64] Cadmium is otherwise stable in air and in water, at ambient conditions, protected by a layer of cadmium oxide.
Mercury is a liquid at room temperature. It has the weakest metallic bonding of all, as indicated by its bonding energy (61 kJ/mol) and melting point (−39 °C) which, together, are the lowest of all the metallic elements.[68][n 8] Solid mercury (MH 1.5)[69] has a distorted crystalline structure,[70] with mixed metallic-covalent bonding,[71] and a BCN of 6. “All of the [Group 12] metals, but especially mercury, tend to form covalent rather than ionic compounds.”[72] The oxide of mercury in its preferred oxidation state (HgO; +2) is weakly amphoteric, as is the congener sulfide HgS.[73] It forms anionic thiomercurates (such as Na2HgS2 and BaHgS3) in strongly basic solutions.[74][n 9] It forms or is a part of Zintl phases such as NaHg and K8In10Hg.[75] Mercury is a relatively inert metal, showing little oxide formation at room temperature.[76]
Group 13[edit]
Main article: Boron group

Aluminium

Gallium

Indium

Thallium
Aluminium sometimes is[77] or is not[3] counted as a post-transition metal. It has a well shielded [Ne] noble gas core rather than the less well shielded [Ar]3d10, [Kr]4d10 or [Xe]4f145d10 core of the post-transition metals. The small radius of the aluminium ion combined with its high charge make it a strongly polarizing species, prone to covalency.[78]
Aluminium in pure form is a soft metal (MH 3.0) with low mechanical strength.[79] It has a close-packed structure (BCN 12) showing some evidence of partially directional bonding.[80][n 10] It has a low melting point and a high thermal conductivity. Its strength is halved at 200 °C, and for many of its alloys is minimal at 300 °C.[82] The latter three properties of aluminium limit its use to situations where fire protection is not required,[83] or necessitate the provision of increased fire protection.[84][n 11] It bonds covalently in most of its compounds;[88] has an amphoteric oxide; and can form anionic aluminates.[60] Aluminium forms Zintl phases such as LiAl, Ca3Al2Sb6, and SrAl2.[89] A thin protective layer of oxide confers a reasonable degree of corrosion resistance.[90] It is susceptible to attack in low pH (<4) and high (> 8.5) pH conditions,[91][n 12] a phenomenon that is generally more pronounced in the case of commercial purity aluminium and aluminium alloys.[97] Given many of these properties and its proximity to the dividing line between metals and nonmetals, aluminium is occasionally classified as a metalloid.[n 13] Despite its shortcomings, it has a good strength-to-weight ratio and excellent ductility; its mechanical strength can be improved considerably with the use of alloying additives; its very high thermal conductivity can be put to good use inheat sinks and heat exchangers;[98] and it has a high electrical conductivity.[n 14] At lower temperatures, aluminium increases its deformation strength (as do most materials) whilst maintaining ductility (as do face-centred cubic metals generally).[100] Chemically, bulk aluminium is a strongly electropositive metal, with a high negative electrode potential.[101][n 15]
Gallium is a soft, brittle metal (MH 1.5) that melts at only a few degrees above room temperature.[103] It has an unusual crystalline structure featuring mixed metallic-covalent bonding and low symmetry[103] (BCN 7 i.e. 1+2+2+2).[104] It bonds covalently in most of its compounds,[105] has an amphoteric oxide;[106] and can form anionic gallates.[60] Gallium forms Zintl phases such as Li2Ga7, K3Ga13 and YbGa2.[107] It is slowly oxidized in moist air at ambient conditions; a protective film of oxide prevents further corrosion.[108]
Indium is a soft, highly ductile metal (MH 1.0) with a low tensile strength.[109][110] It has a partially distorted crystalline structure (BCN 4+8) associated with incompletely ionised atoms.[111] The tendency of indium ‘…to form covalent compounds is one of the more important properties influencing its electrochemical behavior’.[112] The oxides of indium in its preferred oxidation state of +3, namely In2O3 and In(OH)3 are weakly amphoteric; it forms anionic indates in strongly basic solutions.[113] Indium forms Zintl phases such as LiIn, Na2In and Rb2In3.[114] Indium does not oxidize in air at ambient conditions.[110]
Thallium is a soft, reactive metal (MH 1.0), so much so that it has no structural uses.[115] It has a close-packed crystalline structure (BCN 6+6) but an abnormally large interatomic distance that has been attributed to partial ionisation of the thallium atoms.[116] Although compounds in the +1 (mostly ionic) oxidation state are the more numerous, thallium has an appreciable chemistry in the +3 (largely covalent) oxidation state, as seen in its chalcogenides and trihalides.[117] It is the only one of the Group 13 elements to react with air at room temperature, slowly forming the amphoteric oxide Tl2O3.[118][119][120] It forms anionic thallates such as Tl3TlO3, Na3Tl(OH)6, NaTlO2, and KTlO2,[119] and is present as the Tl− thallide anion in the compound CsTl.[121] Thallium forms Zintl phases, such as Na2Tl, Na2K21Tl19, CsTl and Sr5Tl3H.[122]
Group 14[edit]
Main article: Carbon group

Germanium

Tin

Lead
Germanium is a hard (MH 6), very brittle semi-metallic element.[123] It was originally thought to be a poorly conducting metal[124] but has the electronic band structure of a semiconductor.[125] Germanium is usually considered to be a metalloid rather than a metal.[126] Like carbon (as diamond) and silicon, it has a covalent tetrahedral crystalline structure (BCN 4).[127] Compounds in its preferred oxidation state of +4 are covalent.[128] Germanium forms an amphoteric oxide, GeO2[129] and anionic germanates, such as Mg2GeO4.[130] It forms Zintl phases such as LiGe, K8Ge44 and La4Ge3.[131]
Tin is a soft, exceptionally[132] weak metal (MH 1.5);[n 16] a 1-cm thick rod will bend easily under mild finger pressure.[132] It has an irregularly coordinated crystalline structure (BCN 4+2) associated with incompletely ionised atoms.[111] All of the Group 14 elements form compounds in which they are in the +4, predominantly covalent, oxidation state; even in the +2 oxidation state tin generally forms covalent bonds.[134] The oxides of tin in its preferred oxidation state of +2, namely SnO and Sn(OH)2, are amphoteric;[135] it forms stannites in strongly basic solutions.[60]Below 13 °C (55.4 °F) tin changes its structure and becomes ‘grey tin’, which has the same structure as diamond, silicon and germanium (BCN 4). This transformation causes ordinary tin to crumble and disintegrate since, as well as being brittle, grey tin occupies more volume due to having a less efficient crystalline packing structure. Tin forms Zintl phases such as Na4Sn, BaSn, K8Sn25 and Ca31Sn20.[136] It has good corrosion resistance in air on account of forming a thin protective oxide layer. Pure tin has no structural uses.[137] It is used in lead-free solders, and as a hardening agent in alloys of other metals, such as copper, lead, titanium and zinc.[138]
Lead is a soft metal (MH 1.5) which, in many cases,[139] is unable to support its own weight.[140] It has a close-packed structure (BCN 12) but an abnormally large inter-atomic distance that has been attributed to partial ionisation of the lead atoms.[116][141] It forms a semi-covalent dioxide PbO2; a covalently bonded sulfide PbS; covalently bonded halides;[142] and a range of covalently bonded organolead compounds such as the lead(II) mercaptan Pb(SC2H5)2, lead tetra-acetate Pb(CH3CO2)4, and the once common, anti-knock additive, tetra-ethyl lead (CH3CH2)4Pb.[143] The oxide of lead in its preferred oxidation state (PbO; +2) is amphoteric;[144] it forms anionic plumbates in strongly basic solutions.[60] Lead forms Zintl phases such as CsPb, Sr31Pb20, La5Pb3N and Yb3Pb20.[145] It has reasonable to good corrosion resistance; in moist air it forms a mixed gray coating of oxide, carbonate and sulfate that hinders further oxidation.[146]
Group 15[edit]
Main article: Pnictogen

Arsenic

Antimony

Bismuth
Arsenic is a moderately hard (MH 3.5) and brittle semi-metallic element. It is commonly regarded as a metalloid, or by some other authors as either a metal or a non-metal. It exhibits poor electrical conductivity which, like a metal, decreases with temperature. It has a relatively open and partially covalent crystalline structure (BCN 3+3). Arsenic forms covalent bonds with most other elements. The oxide in its preferred oxidation state (As2O3, +3) is amphoteric,[n 17] as is the corresponding oxoacid in aqueous solution (H3AsO3) and congener sulphide (As2S3). Arsenic forms a series of anionic arsenates such as Na3AsO3and PbHAsO4, and Zintl phases such as Na3As, Ca2As and SrAs3.
Antimony is a soft (MH 3.0) and brittle semi-metallic element. It is commonly regarded as a metalloid, or by some other authors as either a metal or a non-metal. It exhibits poor electrical conductivity which, like a metal, decreases with temperature. It has a relatively open and partially covalent crystalline structure (BCN 3+3). Antimony forms covalent bonds with most other elements. The oxide in its preferred oxidation state (Sb2O3, +3) is amphoteric. Antimony forms a series of anionic antimonites and antimonates such as NaSbO2 and AlSbO4, and Zintl phases such as K5Sb4, Sr2Sb3 and BaSb3.
Bismuth is a slightly radioactive, soft metal (MH 2.5) that is too brittle for any structural use.[149] It has an open-packed crystalline structure (BCN 3+3) with bonding that is intermediate between metallic and covalent.[150] For a metal, it has exceptionally low electrical and thermal conductivity.[151] Most of the ordinary compounds of bismuth are covalent in nature.[152] The oxide, Bi2O3 is predominantly basic but will act as a weak acid in warm, very concentrated KOH.[153] It can also be fused with potassium hydroxide in air, resulting in a brown mass of potassium bismuthate.[154] The solution chemistry of bismuth is characterised by the formation of oxyanions;[155] it forms anionic bismuthates in strongly basic solutions.[156] Bismuth forms Zintl phases such as NaBi,[157] Rb7In4Bi6[158] and Ba11Cd8Bi14.[159] Bailar et al.[160] refer to bismuth as being, ‘the least “metallic” metal in its physical properties’ given its brittle nature (and possibly) ‘the lowest electrical conductivity of all metals.’[n 18]
Group 16[edit]
Main article: Polonium
Polonium is a radioactive, soft metal with a hardness similar to lead.[162] It has a simple cublc crystalline structure characterised (as determined by electron density calculations) by partially directional bonding,[163] and a BCN of 6. Such a structure ordinarily results in very low ductility and fracture resistance[164] however polonium has been predicted to be a ductile metal.[165] It forms a covalent hydride;[166] its halides are covalent, volatile compounds, resembling those of tellurium.[167] The oxide of polonium in its preferred oxidation state (PoO2; +4) is predominantly basic, but amphoteric if dissolved in concentrated aqueous alkali, or fused with potassium hydroxide in air.[168] The yellow polonate(IV) ionPoO2−
3 is known in aqueous solutions of low Cl‒ concentration and high pH.[169][n 19] Polonides such as Na2Po, BePo, ZnPo, CdPo and HgPo feature Po2− anions;[171] except for HgPo these are some of the more stable of the polonium compounds.[172][n 20]
Group 17[edit]
Main article: Astatine
Astatine is a radioactive element that has never been seen; a visible quantity would immediately be vapourized due to its intense radioactivity.[174] It may be possible to prevent this with sufficient cooling.[175] Astatine is commonly regarded as a nonmetal,[176] less commonly as a metalloid[177] and occasionally as a metal. Unlike its lighter congener iodine, evidence for diatomic astatine is sparse and inconclusive.[178] In 2013, on the basis of relativistic modelling, astatine was predicted to be a monatomic metal, with a face-centered cubic crystalline structure.[175] As such, astatine could be expected to have a metallic appearance; show metallic conductivity; and have excellent ductility, even at cryogenic temperatures.[179] It could also be expected to show significant nonmetallic character, as is normally the case for metals in, or in the vicinity of, the p-block. Astatine oxyanions AtO−, AtO−
2, AtO−
3 and AtO−
4 are known,[180] oxyanion formation being a tendency of nonmetals.[181] The hydroxide of astatine At(OH) is presumed to be amphoteric.[182][n 21]Astatine forms covalent compounds with nonmetals,[185] including hydrogen astatide HAt and carbon tetraastaide CAt4.[186][n 22] At− anions have been reported to form astatides with silver, thallium, palladium and lead.[188] Pruszyński et al. note that astatide ions should form strong complexes with soft metal cations such as Hg2+, Pd2+, Ag+ and Tl3+; they list the astatide formed with mercury as Hg(OH)At.[189]
Related groupings[edit]
B-subgroup metals[edit]
Superficially, the B-subgroup metals are the metals in Groups IB to VIB of the periodic table, corresponding to Groups 11 to 16 using current IUPAC nonmenclature. Practically, the group 11 metals (copper, silver and gold) are ordinarily regarded as transition metals (or sometimes as coinage metals, or noble metals) whereas the group 12 metals (zinc, cadmium, and mercury) may or may not be treated as B-subgroup metals depending on if the transition metals are taken to end at group 11 or group 12. The ‘B’ nomenclature (as in Groups IB, IIB, and so on) was superseded in 1988 but is still occasionally encountered in more recent literature.[190][n 23]
The B-subgroup metals show nonmetallic properties; this is particularly apparent in moving from group 12 to group 16.[192] Although the group 11 metals have normal close-packed metallic structures[193] they show an overlap in chemical properties. In their +1 compounds (the stable state for silver; less so for copper)[194] they are typical B-subgroup metals. In their +2 and +3 states their chemistry is typical of transition metal compounds.[195]
Borderline metals[edit]
Parish[196] writes that, ‘as anticipated’, the borderline metals of groups 13 and 14 have non-standard structures. Gallium, indium, thallium, germanium, and tin are specifically mentioned in this context. The group 12 metals are also noted as having slightly distorted structures; this has been interpreted as evidence of weak directional (i.e. covalent) bonding.[n 24]
Chemically weak metals[edit]
Rayner-Canham and Overton[198] use the term chemically weak metals to refer to the metals close to the metal-nonmetal borderline. These metals behave chemically more like the metalloids, particularly with respect to anionic species formation. The nine chemically weak metals identified by them are beryllium, aluminium, zinc, gallium, tin, lead, antimony, bismuth, and polonium.[n 25]
Fusible metals[edit]
Cardarelli,[200] writing in 2008, categorizes zinc, cadmium, mercury, gallium, indium thallium, tin, lead, antimony and bismuth as fusible metals. Nearly 100 years earlier, Louis (1911)[201] noted that fusible metals were alloys containing tin, cadmium, lead, and bismuth in various proportions, “the tin ranging from 10 to 20%.”
Heavy metals (of low melting point)[edit]
Van Wert[202] grouped the periodic table metals into a. the light metals; b. the heavy brittle metals of high melting point, c. the heavy ductile metals of high melting point; d. the heavy metals of low melting point (Zn, Cd, Hg; Ga, In, Tl; Ge, Sn; As, Sb, Bi; and Po), and e. the strong, electropositive metals. Britton, Abbatiello and Robins[203] speak of ‘the soft, low melting point, heavy metals in columns lIB, IlIA, IVA, and VA of the periodic table, namely Zn, Cd, Hg; Al, Ga, In, Tl; [Si], Ge, Sn, Pb; and Bi. The Sargent-Welch Chart of the Elements groups the metals into: light metals, the lanthanide series; the actinide series; heavy metals (brittle); heavy metals (ductile); and heavy metals (low melting point): Zn, Cd, Hg, [Cn]; Al, Ga, In, Tl; Ge, Sn, Pb, [Fl]; Sb, Bi; and Po.[204][n 26]
Less typical metals[edit]
Habashi[206] groups the elements into eight major categories: [1] typical metals (alkali metals, alkaline earth metals, and aluminium); [2] lanthanides (Ce–Lu); [3] actinides (Th–Lr); [4] transition metals (Sc, Y, La, Ac, groups 4–10); [5] less typical metals (groups 11–12, Ga, In, Tl, Sn and Pb); [6] metalloids (B, Si, Ge, As, Se, Sb, Te, Bi and Po); [7] covalent nonmetals (H, C, N, O, P, S and the halogens); and [8] monatomic nonmetals (that is, the noble gases).
Metametals[edit]
The metametals are zinc, cadmium, mercury, indium, thallium, tin and lead. They are ductile elements but, compared to their metallic periodic table neighbours to the left, have lower melting points, relatively low electrical and thermal conductivities, and show distortions from close-packed forms.[207] Sometimes beryllium[208] and gallium[209] are included as metametals despite having low ductility.
Ordinary metals[edit]
Abrikosov[210] distinguishes between ordinary metals, and transition metals where the inner shells are not filled. The ordinary metals have lower melting points and cohesive energies than those of the transition metals.[211] Gray[212] identifies as ordinary metals: aluminium, gallium, indium, thallium, element 113, tin, lead, [flerovium], bismuth, element 115, and [livermorium]. He adds that, ‘in reality most of the metals that people think of as ordinary are in fact transition metals…’.
Other metals[edit]
As noted, the metals falling between the transition metals and the metalloids on the periodic table are sometimes called other metals (see also, for example, Taylor et al.).[213]‘Other’ in this sense has the related meanings of, ‘existing besides, or distinct from, that already mentioned’[214] (that is, the alkali and alkaline earth metals, the lanthanides and actinides, and the transition metals); ‘auxiliary’; ‘ancillary, secondary’.[215] According to Gray[216] there should be a better name for these elements than ‘other metals’.
P-block metals[edit]
The p-block metals are the metals in groups 13‒15 (or 16) of the periodic table. Usually, this includes aluminium, gallium, indium and thallium; tin and lead; and bismuth. Germanium, antimony and polonium are sometimes also included, although the first two are commonly recognised as metalloids. The p-block metals tend to have structures that display low coordination numbers and directional bonding. Pronounced covalency is found in their compounds; the majority of their oxides are amphoteric.[217]
Peculiar metals[edit]
Slater[218] divides the metals ‘fairly definitely, though not perfectly sharply’ into the ordinary metals and the peculiar metals, the latter of which verge on the nonmetals. The peculiar metals occur towards the ends of the rows of the periodic table and include ‘approximately:’ gallium, indium, and thallium; carbon, silicon ‘(both of which have some metallic properties, though we have previously treated them as nonmetals),’ germanium and tin; arsenic, antimony, and bismuth; and selenium ‘(which is partly metallic)’ and tellurium. The ordinary metals have centro-symmetrical crystalline structures[n 27] whereas the peculiar metals have structures involving directional bonding. More recently, Joshua observed that the peculiar metals have mixed metallic-covalent bonding.[220]
Poor metals[edit]
Farrell and Van Sicien[221] use the term poor metal, for simplicity, ‘to denote one with a significant covalent, or directional character.’ Hill and Holman[222] observe that, ‘The term poor metals is not widely used, but it is a useful description for several metals including tin, lead and bismuth. These metals fall in a triangular block of the periodic table to the right of the transition metals. They are usually low in the activity (electrochemical) series and they have some resemblances to non-metals.’ Reid et al.[223] write that ‘poor metals’ is, ‘[A]n older term for metallic elements in Groups 13‒15 of the periodic table that are softer and have lower melting points than the metals traditionally used for tools.’
Semimetals[edit]
In modern use, the term ‘semimetal’ sometimes refers, loosely or explicitly, to metals with incomplete metallic character in crystalline structure, electrical conductivity or electronic structure. Examples include gallium,[224] ytterbium,[225] bismuth,[226] mercury[227] and neptunium.[228] Metalloids, which are in-between elements that are neither metals nor nonmetals, are also sometimes instead called semimetals. The elements commonly recognised as metalloids are boron, silicon, germanium, arsenic, antimony and tellurium. In old chemistry, before the publication in 1789 of Lavoisier’s ‘revolutionary’[229] Elementary Treatise on Chemistry,[230] a semimetal was a metallic element with ‘very imperfect ductility and malleability’[231] such as zinc, mercury or bismuth.
Transition metals[edit]
Historically, the transition metal series “includes those elements of the Periodic Table which ‘bridge the gap’ between the very electropositive alkali and allkaline earth metals and the electronegative non-metals of the groups: nitrogen-phosphorus, oxygen-sulfur, and the halogens.”[232] Cheronis, Parsons and Ronneberg[233] wrote that, “The transition metals of low melting point form a block in the Periodic Table: those of Groups II ‘b’ [zinc, cadmium, mercury], III ‘b’ [aluminium, gallium, indium, thallium], and germanium, tin and lead in Group IV. These metals all have melting points below 425 °C.”[n 28]
Transition metal
From Wikipedia, the free encyclopedia
|
|
This article may require cleanup to meet Wikipedia’s quality standards. The specific problem is: Not completely neutral, illogical and self-contradictory at times, and incomplete. Full reasons on talk page. (August 2013) |
| Transition metals in the periodic table | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In chemistry, the term transition metal (or transition element) has two possible meanings:
- The IUPAC definition[1] defines a transition metal as “an element whose atom has a partially filled d sub-shell, or which can give rise to cations with an incomplete d sub-shell”.
- Most scientists describe a “transition metal” as any element in the d-block of the periodic table, which includes groups 3 to 12 on the periodic table.[2][3] In actual practice, the f-block lanthanide and actinide series are also considered transition metals and are called “inner transition metals”.
English chemist Charles Bury first used the word transition in 1921 when he referred to a transition series of elements during the change of an inner layer of electrons (for example n=3 in the 4th row of the periodic table) from a stable group of 8 to one of 18, or from 18 to 32.[4][5] These elements are now known as the d-block.
Classification[edit]
In the d-block the atoms of the elements have between 1 and 10 d electrons.
| Transition metals in the d-block | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Period 4 | Sc 21 | Ti 22 | V 23 | Cr 24 | Mn 25 | Fe 26 | Co 27 | Ni 28 | Cu 29 | Zn 30 |
| Period 5 | Y 39 | Zr 40 | Nb 41 | Mo 42 | Tc 43 | Ru 44 | Rh 45 | Pd 46 | Ag 47 | Cd 48 |
| Period 6 | 57–71 | Hf 72 | Ta 73 | W 74 | Re 75 | Os 76 | Ir 77 | Pt 78 | Au 79 | Hg 80 |
| Period 7 | 89–103 | Rf 104 | Db 105 | Sg 106 | Bh 107 | Hs 108 | Mt 109 | Ds 110 | Rg 111 | Cn 112 |
The typical electronic structure of transition metal atoms can be written as [ ]ns2(n-1)dm, following the Madelung rule where the inner d orbital is predicted to be filled after thevalence-shell s orbital. This is actually not the case; the 4s electrons are higher in energy than the 3d as shown spectroscopically. An ion such as Fe2+
has no 4s electrons: it has the electronic configuration [Ar]3d6 as compared with the configuration of the atom, [Ar]4s23d6.
The elements of groups 3–12 are now generally recognized as transition metals, although the elements La-Lu and Ac-Lr and Group 12 attract different definitions from different authors.
- Many chemistry textbooks and printed periodic tables classify La and Ac as Group 3 elements and transition metals, since their atomic ground-state configurations are s2d1 like Sc and Y. The elements Ce-Lu are considered as the “lanthanide” series (or “lanthanoid” according to IUPAC) and Th-Lr as the “actinide” series.[6][7] The two series together are classified as f-block elements, or (in older sources) as “inner transition elements”.
- Some inorganic chemistry textbooks include La with the lanthanides and Ac with the actinides.[8][9][10] This classification is based on similarities in chemical behaviour, and defines 15 elements in each of the two series even though they correspond to the filling of an f subshell which can only contain 14 electrons.
- A third classification defines the f-block elements as La-Yb and Ac-No, while placing Lu and Lr in Group 3.[4] This is based on the aufbau principle (or Madelung rule) for filling electron subshells, in which 4f is filled before 5d (and 5f before 6d), so that the f subshell is actually full at Yb (and No) while Lu (and Lr) has an [ ]s2f14d1configuration. However La and Ac are exceptions to the Aufbau principle with electron configuration [ ]s2d1 (not [ ]s2f1 as the aufbau principle predicts) so it is not clear from atomic electron configurations whether La or Lu (Ac or Lr) should be considered as transition metals. Eric Scerri has proposed placing Lu and Lr in group 3 on the grounds of continuous sequences of atomic numbers in an expanded or long-form periodic table.[11]
Zinc, cadmium, and mercury are sometimes excluded from the transition metals[4] as they have the electronic configuration [ ]d10s2, with no incomplete d shell.[12] In the oxidation state +2 the ions have the electronic configuration [ ] d10. However, these elements can exist in other oxidation states, including the +1 oxidation state, as in the diatomic ionHg2+
2. The group 12 elements Zn, Cd and Hg may be classed as post-transition metals in this case, because of the formation of a covalent bond between the two atoms of the dimer. However, it is often convenient to include these elements in a discussion of the transition elements. For example, when discussing the crystal field stabilization energy of first-row transition elements, it is convenient to also include the elements calcium and zinc, as both Ca2+
and Zn2+
have a value of zero against which the value for other transition metal ions may be compared. Another example occurs in the Irving-Williams series of stability constants of complexes.
The recent synthesis of mercury(IV) fluoride (HgF
4) has been taken by some to reinforce the view that the group 12 elements should be considered transition metals,[13] but some authors still consider this compound to be exceptional.[14]
Position in the periodic table[edit]
The d-block as stated earlier, is present in the centre of the long form of periodic table. These are flanked or surrounded by elements belonging to s and p-blocks on both sides. These are called transition elements since they represent a transition i.e., there is a change from metallic character of s-block elements to non-metallic character of p-block elements through d-block elements which are also metals. As pointed above there are four transition series in this block. Since the filling of electrons takes place in (n-1)d orbitals, the periods to which these series belong, is actually one more than the actual series. For example, the elements included in 3d series belong to fourth period ; the elements included in 4d series belong to the fifth period and so on.
Electronic configuration[edit]
The general electronic configuration of the d-block elements is [Inert gas] (n-1)d1–10n s1–2. The period 6 and 7 transition metals also add (n-2)f14 electrons, which are omitted from the tables below.
The d-sub-shell is the penultimate (last but one) sub-shell and is denoted as (n-1) d-sub-shell. The number of s electrons may vary from one to two. The s-sub-shell in the valence shell is represented as the ns sub-shell. However, palladium (Pd) is an exception with no electron in the s-sub shell. In the periodic table, the transition metals are present in ten groups (3 to 12). Group-2 belongs to the s– block with an ns2 configuration.
The elements in group-3 have an ns2(n-1)d1 configuration. The first transition series is present in the 4thperiod, and starts after Ca (Z=20) of group-2 which has configuration [Ar]4s2. The electronic configuration of scandium (Sc), the first element of group-3 with atomic number Z=21 is[Ar]4s23d1. As we move from left to right, electrons are added to the same d-sub-shell till it is complete. The element of group-12 in the first transition series is zinc (Zn) with configuration [Ar]4s23d10. Since the electrons added fill the (n-1)dorbitals, the properties of the d-block elements are quite different from those of s and p block elements in which the filling occurs either in s or in p-orbitals of the valence shell. The electronic configuration of the individual elements present in all the transition series are given below:
First (3d) Transition Series (Sc-Zn)
| Group | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| At.no. | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 |
| Element | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn |
| Config. | 3d14s2 | 3d24s2 | 3d34s2 | 3d54s1 | 3d54s2 | 3d64s2 | 3d74s2 | 3d84s2 | 3d104s1 | 3d104s2 |
Second (4d) Transition Series (Y-Cd)
| At. No. | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 47 | 48 |
| Element | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd |
| Config. | 4d15s2 | 4d25s2 | 4d45s1 | 4d55s1 | 4d55s2 | 4d75s1 | 4d85s1 | 4d105s0 | 4d105s1 | 4d105s2 |
Third (5d) Transition Series (Lu-Hg)[15]
| At.No | 71 | 72 | 73 | 74 | 75 | 76 | 77 | 78 | 79 | 80 |
| Element | Lu | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg |
| Config. | 5d16s2 | 5d26s2 | 5d36s2 | 5d46s2 | 5d56s2 | 5d66s2 | 5d76s2 | 5d96s1 | 5d106s1 | 5d106s2 |
Fourth (6d) Transition Series (Lr-Cn)
| At. No. | 103 | 104 | 105 | 106 | 107 | 108 | 109 | 110 | 111 | 112 |
| Element | Lr | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | Cn |
| Config. | 7s27p1 | 6d27s2 | 6d37s2 | 6d47s2 | 6d57s2 | 6d67s2 | 6d77s2 | 6d87s2 | 6d97s2 | 6d107s2 |
A careful look at the electronic configuration of the elements reveals that there are certain exceptions shown by Pt, Au and Hg.. These are either because of the symmetry or nuclear-electron and electron-electron force.
The (n-1)d orbitals that are involved in the transition metals are very significant because they influence such properties as magnetic character, variable oxidation states, formation of colored compounds etc. The valence s(ns) and p(np) orbitals have very little contribution in this regard since they hardly change in the moving from left to the right in a transition series. In transition metals, there is a greater horizontal similarities in the properties of the elements in a period in comparison to the periods in which the d-orbitals are not involved. This is because in a transition series, the valence shell electronic configuration of the elements do not change. However, there are some group similarities as well.
Characteristic properties[edit]
There are a number of properties shared by the transition elements that are not found in other elements, which results from the partially filled d shell. These include
- the formation of compounds whose colour is due to d–d electronic transitions
- the formation of compounds in many oxidation states, due to the relatively low reactivity of unpaired d electrons.[16]
- the formation of many paramagnetic compounds due to the presence of unpaired d electrons. A few compounds of main group elements are also paramagnetic (e.g. nitric oxide, oxygen)
Coloured compounds[edit]
From left to right, aqueous solutions of:Co(NO
3)
2 (red); K
2Cr
2O
7 (orange); K
2CrO
4(yellow); NiCl
2 (turquoise); CuSO
4 (blue);KMnO
4 (purple).
Colour in transition-series metal compounds is generally due to electronic transitions of two principal types.
- charge transfer transitions. An electron may jump from a predominantly ligand orbital to a predominantly metal orbital, giving rise to a ligand-to-metal charge-transfer (LMCT) transition. These can most easily occur when the metal is in a high oxidation state. For example, the colour of chromate, dichromate and permanganate ions is due to LMCT transitions. Another example is thatmercuric iodide, HgI2, is red because of a LMCT transition.
A metal-to-ligand charge transfer (MLCT) transition will be most likely when the metal is in a low oxidation state and the ligand is easily reduced.
- d–d transitions. An electron jumps from one d-orbital to another. In complexes of the transition metals the d orbitals do not all have the same energy. The pattern of splitting of the d orbitals can be calculated using crystal field theory. The extent of the splitting depends on the particular metal, its oxidation state and the nature of the ligands. The actual energy levels are shown on Tanabe-Sugano diagrams.
In centrosymmetric complexes, such as octahedral complexes, d–d transitions are forbidden by the Laporte rule and only occur because of vibronic coupling in which a molecular vibration occurs together with a d-d transition. Tetrahedral complexes have somewhat more intense colour because mixing d and p orbitals is possible when there is no centre of symmetry, so transitions are not pure d-d transitions. The molar absorptivity (ε) of bands caused by d-d transitions are relatively low, roughly in the range 5-500 M−1cm−1 (whereM = mol dm−3).[17] Some d–d transitions are spin forbidden. An example occurs in octahedral, high-spin complexes of manganese(II), which has a d5 configuration in which all five electron has parallel spins; the colour of such complexes is much weaker than in complexes with spin-allowed transitions. Many compounds of manganese(II) appear almost colourless. The spectrum of [Mn(H
2O)
6]2+
shows a maximum molar absorptivity of about 0.04 M−1cm−1 in the visible spectrum.
Oxidation states[edit]
A characteristic of transition metals is that they exhibit two or more oxidation states, usually differing by one. For example, compounds of vanadium are known in all oxidation states between −1, such as [V(CO)
6]−
, and +5, such as VO3−
4.
Main group elements in groups 13 to 17 also exhibit multiple oxidation states. The “common” oxidation states of these elements typically differ by two. For example, compounds of gallium in oxidation states +1 and +3 exist in which there is a single gallium atom. No compound of Ga(II) is known: any such compound would have an unpaired electron and would behave as a free radical and be destroyed rapidly. The only compounds in which gallium has a formal oxidation state of +2 are dimeric compounds, such as [Ga
2Cl
6]2−
, which contain a Ga-Ga bond formed from the unpaired electron on each Ga atom.[18] Thus the main difference in oxidation states, between transition elements and other elements is that oxidation states are known in which there is a single atom of the element and one or more unpaired electrons.
The maximum oxidation state in the first row transition metals is equal to the number of valence electrons from titanium (+4) up to manganese (+7), but decreases in the later elements. In the second row the maximum occurs with ruthenium (+8), and in the third row, the maximum occurs with iridium (+9). In compounds such as [MnO
4]−
and OsO
4 the elements achieve a stable octet by forming four covalent bonds.
The lowest oxidation states are exhibited in metal carbonyl complexes such as Cr(CO)
6 (oxidation state zero) and [Fe(CO)
4]2−
(oxidation state −2) in which the 18-electron rule is obeyed. These complexes are also covalent.
Ionic compounds are mostly formed with oxidation states +2 and +3. In aqueous solution the ions are hydrated by (usually) six water molecules arranged octahedrally.
Magnetism[edit]
Main article: magnetochemistry
Transition metal compounds are paramagnetic when they have one or more unpaired d electrons.[19] In octahedral complexes with between four and seven d electrons both high spin and low spin states are possible. Tetrahedral transition metal complexes such as [FeCl
4]2−
are high spin because the crystal field splitting is small so that the energy to be gained by virtue of the electrons being in lower energy orbitals is always less than the energy needed to pair up the spins. Some compounds are diamagnetic. These include octahedral, low-spin, d6 and square-planar d8 complexes. In these cases, crystal field splitting is such that all the electrons are paired up.
Ferromagnetism occurs when individual atoms are paramagnetic and the spin vectors are aligned parallel to each other in a crystalline material. Metallic iron and the alloy alnicoare examples of ferromagnetic materials involving transition metals. Anti-ferromagnetism is another example of a magnetic property arising from a particular alignment of individual spins in the solid state.
Catalytic properties[edit]
The transition metals and their compounds are known for their homogeneous and heterogeneous catalytic activity. This activity is ascribed to their ability to adopt multiple oxidation states and to form complexes. Vanadium(V) oxide (in the contact process), finely divided iron (in the Haber process), and nickel (in catalytic hydrogenation) are some of the examples. Catalysts at a solid surface (nanomaterial-based catalysts) involve the formation of bonds between reactant molecules and atoms of the surface of the catalyst (first row transition metals utilize 3d and 4s electrons for bonding). This has the effect of increasing the concentration of the reactants at the catalyst surface and also weakening of the bonds in the reacting molecules (the activation energy is lowered). Also because the transition metal ions can change their oxidation states, they become more effective ascatalysts.
Other properties[edit]
As implied by the name, all transition metals are metals and conductors of electricity.
In general, transition metals possess a high density and high melting points and boiling points. These properties are due to metallic bonding by delocalized d electrons, leading tocohesion which increases with the number of shared electrons. However the group 12 metals have much lower melting and boiling points since their full d subshells prevent d–d bonding. Mercury has a melting point of −38.83 °C (−37.89 °F) and is a liquid at room temperature.
Many transition metals can be bound to a variety of ligands.[20]
See also[edit]
- Inner transition element, a name given to any member of the f-block
- Main group element, an element other than a transition metal.
- Ligand field theory a development of crystal field theory taking covalency into account.
- Crystal field theory a model that describes the breaking of degeneracies of electronic orbital states.
- Post-transition metal, a metallic element to the right of the transition metals in the periodic table.
Alkaline earth metal
From Wikipedia, the free encyclopedia
| Alkaline earth metals | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ↓ Period | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
 Beryllium (Be) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
 Magnesium (Mg) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
 Calcium (Ca) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
 Strontium (Sr) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
 Barium (Ba) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
 Radium (Ra) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Legend
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The alkaline earth metals are six chemical elements in column (group) 2 of the Periodic table. They are beryllium (Be), magnesium(Mg), calcium (Ca), strontium (Sr), barium (Ba), and radium (Ra).[1] They have very similar properties: they are all shiny, silvery-white, somewhat reactive metals at standard temperature and pressure.[2]
Structurally, they have in common an outer s- electron shell which is full;.[2][3][4] that is, this orbital contains its full complement of two electrons, which these elements readily lose to form cations with charge +2, and an oxidation state (oxidation number) of +2.[5]
All the discovered alkaline earth metals occur in nature.[6] Experiments have been conducted to attempt the synthesis of element 120, the next potential member of the group, but they have all met with failure.
Characteristics[edit]
Chemical[edit]
As with other groups, the members of this family show patterns in their electronic configuration, especially the outermost shells, resulting in trends in chemical behavior:
| Z | Element | No. of electrons/shell | Electron configuration[n 1] |
|---|---|---|---|
| 4 | beryllium | 2, 2 | [He] 2s2 |
| 12 | magnesium | 2, 8, 2 | [Ne] 3s2 |
| 20 | calcium | 2, 8, 8, 2 | [Ar] 4s2 |
| 38 | strontium | 2, 8, 18, 8, 2 | [Kr] 5s2 |
| 56 | barium | 2, 8, 18, 18, 8, 2 | [Xe] 6s2 |
| 88 | radium | 2, 8, 18, 32, 18, 8, 2 | [Rn] 7s2 |
Most of the chemistry has been observed only for the first five members of the group. The chemistry of radium is not well-established due to its radioactivity;[2] thus, the presentation of its properties here is limited.
The alkaline earth metals are all silver-colored and soft, and have relatively low densities, melting points, and boiling points. In chemical terms, all of the alkaline metals react with the halogens to form the alkaline earth metal halides, all of which being ionic crystalline compounds (except for beryllium chloride, which is covalent). All the alkaline earth metals except beryllium also react with water to form strongly alkaline hydroxides and, thus, should be handled with great care. The heavier alkaline earth metals react more vigorously than the lighter ones.[2] The alkaline metals have the second-lowest first ionization energies in their respective periods of the periodic table[4]because of their somewhat low effective nuclear charges and the ability to attain a full outer shell configuration by losing just twoelectrons. The second ionization energy of all of the alkaline metals is also somewhat low.[2][4]
Beryllium is an exception: It does not react with water or steam, and its halides are covalent. If beryllium did form compounds with an ionization state of +2, it would polarize electron clouds that are near it very strongly and would cause extensive orbital overlap, since beryllium has a high charge density. All compounds that include beryllium have a covalent bond.[7] Even the compound beryllium fluoride, which is the most ionic beryllium compound, has a low melting point and a low electrical conductivity when melted.[8][9][10]
All the alkaline earth metals have two electrons in their valence shell, so the energetically preferred state of achieving a filled electron shell is to lose two electrons to form doubly charged positive ions.
Compounds and reactions[edit]
The alkaline earth metals all react with the halogens to form ionic halides, such as calcium chloride (CaCl
2), as well as reacting withoxygen to form oxides such as strontium oxide (SrO). Calcium, strontium, and barium react with water to produce hydrogen gas and their respective hydroxides, and also undergo transmetalation reactions to exchange ligands.
-
Alkaline earth metals fluorides solubility-related constants[n 2] Metal M2+
HE
[11][clarification needed]F−
HE
[12][clarification needed]“MF2”
unit
HEMF2
lattice
energies
[13]Solubility
[14][clarification needed]Be 2,455 458 3,371 3,526 soluble Mg 1,922 458 2,838 2,978 0.0012 Ca 1,577 458 2,493 2,651 0.0002 Sr 1,415 458 2,331 2,513 0.0008 Ba 1,361 458 2,277 2,373 0.006
Physical and atomic[edit]
The table below is a summary of the key physical and atomic properties of the alkaline earth metals.
| Alkaline earth metal | Standard atomic weight (u)[n 3][16][17] |
Melting point (K) |
Melting point (°C) |
Boiling point (K)[4] |
Boiling point (°C)[4] |
Density (g/cm3) |
Electronegativity (Pauling) |
First ionization energy (kJ·mol−1) |
Covalent radius (pm)[18] |
Flame test color | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Beryllium | 9.012182(3) | 1560 | 1287 | 2742 | 2469 | 1.85 | 1.57 | 899.5 | 105 | White[19] | |
| Magnesium | 24.3050(6) | 923 | 650 | 1363 | 1090 | 1.738 | 1.31 | 737.7 | 150 | Brilliant-white[2] | |
| Calcium | 40.078(4) | 1115 | 842 | 1757 | 1484 | 1.54 | 1.00 | 589.8 | 180 | Brick-red[2] | |
| Strontium | 87.62(1) | 1050 | 777 | 1655 | 1382 | 2.64 | 0.95 | 549.5 | 200 | Crimson[2] | |
| Barium | 137.327(7) | 1000 | 727 | 2170 | 1897 | 3.594 | 0.89 | 502.9 | 215 | Apple-green[2] | |
| Radium | [226][n 4] | 973 | 700 | 2010 | 1737 | 5.5 | 0.9 | 509.3 | 221 | Crimson red[n 5] | |
Nuclear stability[edit]
| This section requires expansion.(November 2012) |
All of the alkaline earth metals except magnesium and strontium have at least one naturally occurring radioisotope: beryllium-7, beryllium-10, and calcium-41 are trace radioisotopes, calcium-48 and barium-130 have very long half-lives and, thus, occur naturally, and all isotopes of radium are radioactive. Calcium-48 is the lightest nuclide to undergo double beta decay.[21]
The natural radioisotope of calcium, calcium-48, makes up about 0.1874% of natural calcium,[22] and, thus, natural calcium is weakly radioactive. Barium-130 makes up approximately 0.1062% of natural barium, and, thus, barium is weakly radioactive, as well.[23]
History[edit]
Etymology[edit]
The alkaline earth metals are named after their oxides, the alkaline earths, whose old-fashioned names were beryllia, magnesia, lime, strontia, and baryta. These oxides are basic (alkaline) when combined with water. “Earth” is an old term applied by early chemists to nonmetallic substances that are insoluble in water and resistant to heating—properties shared by these oxides. The realization that these earths were not elements but compounds is attributed to the chemist Antoine Lavoisier. In his Traité Élémentaire de Chimie (Elements of Chemistry) of 1789 he called them salt-forming earth elements. Later, he suggested that the alkaline earths might be metal oxides, but admitted that this was mere conjecture. In 1808, acting on Lavoisier’s idea, Humphry Davy became the first to obtain samples of the metals by electrolysis of their molten earths,[24] thus supporting Lavoisier’s hypothesis and causing the group to be named the alkaline earth metals.
Discovery[edit]
The calcium compounds calcite and lime have been known and used since prehistoric times.[25] The same is true for the beryllium compounds beryl and emerald.[26] The other compounds of the alkaline earth metals were discovered starting in the early 15th century. The magnesium compound magnesium sulfate was first discovered in 1618 by a farmer at Epsom in England. Strontium carbonate was discovered in minerals in the Scottish village of Strontian in 1790. The last element is the least abundant: radioactiveradium, which was extracted from uraninite in 1898.[27][28][29]
All elements except beryllium were isolated by electrolysis of molten compounds. Magnesium, calcium, and strontium were first produced by Humphry Davy in 1808, whereas beryllium was independently isolated by Friedrich Wöhler and Antoine Bussy in 1828 by reacting beryllium compounds with potassium. In 1910, radium was isolated as a pure metal by Curie and André-Louis Debierne also by electrolysis.[27][28][29]
Beryllium[edit]
_15.jpg)
Emerald, a variety of beryl, the mineral that beryllium was first known to be in.
Beryl, a mineral that contains beryllium, has been known since the time of the Ptolemaic dynasty in Egypt.[26] Although it was originally thought that beryl was an aluminium silicate,[30] beryl was later found to contain a then-unknown element when, in 1797, Louis-Nicolas Vauquelin dissolved aluminium hydroxide from beryl in an alkali.[31] In 1828, Friedrich Wöhler[32] and Antoine Bussy[33] independently isolated this new element, beryllium, by the same method, which involved a reaction of beryllium chloride with metallic potassium; this reaction was not able to produce large ingots of beryllium.[34] It was not until 1898, when Paul Lebeau performed an electrolysis of a mixture of beryllium fluoride and sodium fluoride, that large pure samples of beryllium were produced.[34]
Magnesium[edit]
Magnesium was first produced by Sir Humphry Davy in England in 1808 using electrolysis of a mixture of magnesia and mercuric oxide.[35]Antoine Bussy prepared it in coherent form in 1831. Davy’s first suggestion for a name was magnium,[35] but the name magnesium is now used.
Calcium[edit]
Lime has been used as a material for building since 7000 to 14,000 BCE,[25] and kilns used for lime have been dated to 2,500 BCE in Khafaja, Mesopotamia.[36][37] Calcium as a material has been known since at least the first century, as the ancient Romans were known to have used calcium oxide by preparing it from lime. Calcium sulfate has been known to be able to set broken bones since the tenth century. Calcium itself, however, was not isolated until 1808, when Humphry Davy, in England, used electrolysis on a mixture of lime and mercuric oxide,[38] after hearing that Jöns Jakob Berzelius had prepared a calcium amalgam from the electrolysis of lime in mercury.
Strontium[edit]
In 1790, physician Adair Crawford, who had been working with barium, realized that Strontian ores showed different properties than other supposed ores of barium.[39] Therefore, he concluded that these ores contained new minerals, which were named strontites in 1793 by Thomas Charles Hope, a chemistry professor at the University of Glasgow,[40] who confirmed Crawford’s discovery. Strontium was eventually isolated in 1808 by Sir Humphry Davy by electrolysis of a mixture of strontium chloride and mercuric oxide. The discovery was announced by Davy on 30 June 1808 at a lecture to the Royal Society.[41]
Barium[edit]

Barite, the material that was first found to contain barium.
Barite, a mineral containing barium, was first recognized as containing a new element in 1774 by Carl Scheele, although he was able to isolate onlybarium oxide. Barium oxide was isolated again two years later by Johan Gottlieb Gahn. Later in the 18th century, William Withering noticed a heavy mineral in the Cumberland lead mines, which are now known to contain barium. Barium itself was finally isolated in 1808 when Sir Humphry Davyused electrolysis with molten salts, and Davy named the element barium, after baryta. Later, Robert Bunsen and Augustus Matthiessen isolated pure barium by electrolysis of a mixture of barium chloride and ammonium chloride.[42][43]
Radium[edit]
While studying uraninite, on 21 December 1898, Marie and Pierre Curie discovered that, even after uranium had decayed, the material created was still radioactive. The material behaved somewhat similarly to barium compounds, although some properties, such as the color of the flame test and spectral lines, were much different. They announced the discovery of a new element on 26 December 1898 to the French Academy of Sciences.[44]Radium was named in 1899 from the word radius, meaning ray, as radium emitted power in the form of rays.[45]
Occurrence[edit]

Series of alkaline earth metals.
Beryllium occurs in the earth’s crust at a concentration of two to six parts per million (ppm),[46] much of which is in soils, where it has a concentration of six ppm. Beryllium is one of the rarest elements in seawater, even rarer than elements such as scandium, with a concentration of 0.2 parts per trillion.[47][48] However, in freshwater, beryllium is somewhat more common, with a concentration of 0.1 parts per billion.[49]
Magnesium and calcium are very common in the earth’s crust, with calcium the fifth-most-abundant element, and magnesium the eighth. None of the alkaline earth metals are found in their elemental state, but magnesium and calcium are found in many rocks and minerals: magnesium in carnellite, magnesite, and dolomite; and calcium in chalk, limestone, gypsum, and anhydrite.[2]
Strontium is the fifteenth-most-abundant element in the Earth’s crust. Most strontium is found in the minerals celestite and strontianite.[50]Barium is slightly less common, much of it in the mineral barite.[51]
Radium, being a decay product of uranium, is found in all uranium-bearing ores.[52] Due to its relatively short half-life,[53] radium from the Earth’s early history has decayed, and present-day samples have all come from the much slower decay of uranium.[52]
Production[edit]
| This section requires expansion.(November 2012) |

Emerald, a variety of beryl, is a naturally occurring compound of beryllium.
Most beryllium is extracted from beryllium hydroxide. One production method is sintering, done by mixing beryl, sodium fluorosilicate, and soda at high temperatures to form sodium fluoroberyllate, aluminium oxide, and silicon dioxide. A solution of sodium fluoroberyllate and sodium hydroxide in water is then used to form beryllium hydroxide by precipitation. Alternatively, in the melt method, powdered beryl is heated to high temperature, cooled with water, then heated again slightly in sulfuric acid, eventually yielding beryllium hydroxide. The beryllium hydroxide from either method then produces beryllium fluoride and beryllium chloride through a somewhat long process. Electrolysis or heating of these compounds can then produce beryllium.[7]
In general, strontium carbonate is extracted from the mineral celestite through two methods: by leaching the celestite with sodium carbonate, or in a more complicated way involving coal.[54]
To produce barium, barite ore is separated from quartz, sometimes by froth flotation methods, resulting in relatively pure barite. Carbon is then used to reduce the baryte into barium sulfide, which is dissolved with other elements to form other compounds, such as barium nitrate. These in turn are thermally decompressed into barium oxide, which eventually yields pure barium after a reaction with aluminium.[51] The most important supplier of barium is China, which produces more than 50% of world supply.[55]
Applications[edit]
Beryllium is used mostly for military applications,[56] but there are other uses of beryllium, as well. In electronics, beryllium is used as a p-type dopant in some semiconductors,[57]and beryllium oxide is used as a high-strength electrical insulator and heat conductor.[58] Due to its light weight and other properties, beryllium is also used in mechanics when stiffness, light weight, and dimensional stability are required at wide temperature ranges.[59][60]
Magnesium has many different uses. One of its most common uses was in industry, where it has many structural advantages over other materials such as aluminium, although this usage has fallen out of favor recently due to magnesium’s flammability.[61] Magnesium is also often alloyed with aluminium or zinc to form materials with more desirable properties than any pure metal.[62] Magnesium has many other uses in industrial applications, such as having a role in the production of iron and steel, and the production oftitanium.[63]
Calcium also has many uses. One of its uses is as a reducing agent in the separation of other metals from ore, such as uranium. It is also used in the production of the alloys of many metals, such as aluminium and copper alloys, and is also used to deoxidize alloys as well. Calcium also has a role in the making of cheese, mortars, and cement.[64]
Strontium and barium do not have as many applications as the lighter alkaline earth metals, but still have uses. Strontium carbonate is often used in the manufacturing of redfireworks,[65] and pure strontium is used in the study of neurotransmitter release in neurons.[66][67] Barium has some use in vacuum tubes to remove gases,[51] and barium sulfatehas many uses in the petroleum industry,[4] as well as other industries.[4][51][68]
Due to its radioactivity, radium no longer has many applications, but it used to have many. Radium used to be used often in luminous paints,[69] although this use was stopped after workers got sick.[70] As people used to think that radioactivity was a good thing, radium used to be added to drinking water, toothpaste, and many other products, although they are also not used anymore due to their health effects.[61] Radium is no longer even used for its radioactive properties, as there are more powerful and safer emitters than radium.[71][72]
Biological role and precautions[edit]
| This section requires expansion.(January 2013) |
Magnesium and calcium are ubiquitous and essential to all known living organisms. They are involved in more than one role, with, for example, magnesium or calcium ion pumpsplaying a role in some cellular processes, magnesium functioning as the active center in some enzymes, and calcium salts taking a structural role, most notably in bones.
Strontium plays an important role in marine aquatic life, especially hard corals, which use strontium to build their exoskeletons. It and barium have some uses in medicine, for example “barium meals” in radiographic imaging, whilst strontium compounds are employed in some toothpastes. Excessive amounts of strontium-90 are toxic due to its radioactivity and strontium-90 mimics calcium and then can kill.
Beryllium and radium, however, are toxic. Beryllium’s low aqueous solubility means it is rarely available to biological systems; it has no known role in living organisms and, when encountered by them, is usually highly toxic.[7] Radium has a low availability and is highly radioactive, making it toxic to life.
Extensions[edit]
The next alkaline earth metal after radium is thought to be element 120, although this may not be true due to relativistic effects.[73] The synthesis of element 120 was first attempted in March 2007, when a team at the Flerov Laboratory of Nuclear Reactions in Dubna bombarded plutonium-244 with iron-58 ions; however, no atoms were produced, leading to a limit of 400 fb for the cross-section at the energy studied.[74] In April 2007, a team at the GSI attempted to create element 120 by bombarding uranium-238 withnickel-64, although no atoms were detected, leading to a limit of 1.6 pb for the reaction. Synthesis was again attempted at higher sensitivities, although no atoms were detected. Other reactions have been tried, although all have been met with failure.[75]
The chemistry of element 120 is predicted to be closer to that of calcium or strontium[76] instead of barium or radium. This is unusual as periodic trends would predict element 120 to be more reactive than barium and radium. This lowered reactivity is due to the expected energies of element 120’s valence electrons, increasing element 120’s ionization energy and decreasing the metallic and ionic radii.[76]
Alkali metal
From Wikipedia, the free encyclopedia
| Alkali metals | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ↓ Period | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
 Lithium (Li) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
.jpg) Sodium (Na) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
 Potassium (K) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
 Rubidium (Rb) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
 Caesium (Cs) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Francium (Fr) 87 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Legend
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The alkali metals are a group (column) in the periodic table consisting of the chemical elements lithium (Li), sodium (Na), potassium(K),[note 1] rubidium (Rb), caesium (Cs),[note 2] and francium (Fr). This group lies in the s-block of the periodic table of elements as all alkali metals have their outermost electron in an s-orbital: this element/electron configuration results in their characteristic properties. The alkali metals provide the best example of group trends in properties in the periodic table, with elements exhibiting well-characterizedhomologous behaviour.
The alkali metals have very similar properties: they are all shiny, soft, highly reactive metals at standard temperature and pressure and readily lose their outermost electron to form cations with charge +1. They can all be cut easily with a knife due to their softness, exposing a shiny surface that tarnishes rapidly in air due to oxidation by atmospheric moisture and oxygen. Because of their high reactivity, they must be stored under oil to prevent reaction with air, and are found naturally only in salts and never as the free element. In the modernIUPAC nomenclature, the alkali metals comprise the group 1 elements,[note 3] excluding hydrogen (H), which is nominally a group 1 element but not normally considered to be an alkali metal as it rarely exhibits behaviour comparable to that of the alkali metals. All the alkali metals react with water, with the heavier alkali metals reacting more vigorously than the lighter ones.
All of the discovered alkali metals occur in nature: in order of abundance, sodium is the most abundant, followed by potassium, lithium, rubidium, caesium, and finally francium, which is very rare due to its extremely high radioactivity and thus occurs only in traces due to its presence in natural decay chains. Experiments have been conducted to attempt the synthesis of ununennium (Uue), which is likely to be the next member of the group, but they have all met with failure. However, ununennium may not be an alkali metal due to relativistic effects, which are predicted to have a large influence on the chemical properties of superheavy elements; even if it does turn out to be an alkali metal, it is predicted to have some differences in physical and chemical properties from its lighter homologues.
Most alkali metals have many different applications. Two of the most well-known applications of the pure elements are rubidium and caesium atomic clocks, of which caesium atomic clocks are the most accurate and precise representation of time. A common application of the compounds of sodium is the sodium-vapour lamp, which emits very efficient light. Table salt, or sodium chloride, has been used since antiquity. Sodium and potassium are also essential elements, having major biological roles as electrolytes, and although the other alkali metals are not essential, they also have various effects on the body, both beneficial and harmful.
Properties[edit]
Physical and chemical[edit]
The physical and chemical properties of the alkali metals can be readily explained by their having an ns1 valence electron configuration, which results in weak metallic bonding. Hence, all the alkali metals are soft and have low densities,[5] melting[5] and boiling points,[5] as well as heats of sublimation, vaporisation, and dissociation[6]:74 They all crystallize in the body-centered cubic crystal structure,[6]:73 and have distinctive flame colours because their outer s electron is very easily excited.[6]:75 The ns1 configuration also results in the alkali metals having very large atomic and ionic radii, as well as high thermal and electrical conductivity.[6]:75 Their chemistry is dominated by the loss of their lone valence electron to form the +1 oxidation state, due to the ease of ionizing this electron and the very high second ionization energy.[6]:76 Most of the chemistry has been observed only for the first five members of the group. The chemistry of francium is not well established due to its extreme radioactivity;[5] thus, the presentation of its properties here is limited.
| Name | Lithium | Sodium | Potassium | Rubidium | Caesium | Francium |
|---|---|---|---|---|---|---|
| Atomic number | 3 | 11 | 19 | 37 | 55 | 87 |
| Standard atomic weight (u)[note 4][9][10] | 6.94(1)[note 5] | 22.98976928(2) | 39.0983(1) | 85.4678(3) | 132.9054519(2) | [223][note 6] |
| Electron configuration | [He] 2s1 | [Ne] 3s1 | [Ar] 4s1 | [Kr] 5s1 | [Xe] 6s1 | [Rn] 7s1 |
| Melting point | 453.69 K 180.54 °C 356.97 °F |
370.87 K 97.72 °C 207.9 °F |
336.53 K, 63.38 °C, 146.08 °F |
312.467 K, 39.31 °C, 102.76 °F |
301.59 K, 28.44 °C, 83.19 °F |
? 300 K, ? 27 °C, ? 80 °F[note 7] |
| Boiling point | 1615 K, 1342 °C, 2448 °F |
1156 K, 883 °C, 1621 °F |
1032 K, 759 °C, 1398 °F |
961 K, 688 °C, 1270 °F |
944 K, 671 °C, 1240 °F |
? 950 K, ? 677 °C, ? 1250 °F[12][note 7] |
| Density (g·cm−3) | 0.534 | 0.968 | 0.89 | 1.532 | 1.93 | ? 1.87 |
| Heat of fusion (kJ·mol−1) | 3.00 | 2.60 | 2.321 | 2.19 | 2.09 | ? 2 |
| Heat of vaporisation (kJ·mol−1) | 136 | 97.42 | 79.1 | 69 | 66.1 | ? 65 |
| Heat of formation of monatomic gas (kJ·mol−1) | 162 | 108 | 89.6 | 82.0 | 78.2 | ? |
| Electrical resistivity at 298 K (nΩ·cm) | 94.7 | 48.8 | 73.9 | 131 | 208 | ? |
| Atomic radius (pm) | 152 | 186 | 227 | 248 | 265 | ? |
| Ionic radius of M+ ion (pm)[note 8] | 76 | 102 | 138 | 152 | 167 | ? 180 |
| First ionisation energy (kJ·mol−1) | 520.2 | 495.8 | 418.8 | 403.0 | 375.7 | 392.8[13] |
| Electron affinity (kJ·mol−1) | 59.62 | 52.87 | 48.38 | 46.89 | 45.51 | ? 47.2[14] |
| Enthalpy of dissociation of M2 (kJ·mol−1) | 106.5 | 73.6 | 57.3 | 45.6 | 44.77 | ? 42.1[15] |
| Pauling electronegativity | 0.98 | 0.93 | 0.82 | 0.82 | 0.79 | ? 0.7[note 9] |
| Standard electrode potential (E°(M+→M0); V) | −3.0405 | −2.714 | −2.925 | −2.925 | −2.923 | ? |
| Flame test colour Principal emission/absorption wavelength (nm) |
Crimson 670.8 |
Yellow 589.2 |
Violet 766.5 |
Red-violet 780.0 |
Blue 455.5 |
? |
The alkali metals are more similar to each other than the elements in any other group are to each other.[5] For instance, when moving down the table, all known alkali metals show increasing atomic radius,[18] decreasing electronegativity,[18]increasing reactivity,[5] and decreasing melting and boiling points[18] as well as heats of fusion and vaporisation.[6]:75 In general, their densitiesincrease when moving down the table, with the exception that potassium is less dense than sodium.[18] One of the very few properties of the alkali metals that does not display a very smooth trend is their reduction potentials: lithium’s value is anomalous, being more negative than the others.[6]:75 This is because the Li+ ion has a very high hydration energy in the gas phase: though the lithium ion disrupts the structure of water significantly, causing a higher change in entropy, this high hydration energy is enough to make the reduction potentials indicate it as being the most electropositive alkali metal, despite the difficulty of ionizing it in the gas phase.[6]:75
The stable alkali metals are all silver-coloured metals except for caesium, which has a golden tint:[19] it is one of only three metals that are clearly coloured (the other two being copper and gold).[6]:74 Additionally, the heavy alkaline earth metals calcium, strontium, and barium, as well as the divalent lanthanides europium and ytterbium, are pale yellow, though the colour is much less prominent than it is for caesium.[6]:74 Their lustre tarnishes rapidly in air due to oxidation.[5] They all crystallize in the body-centered cubic crystal structure,[6]:73 and have distinctive flame coloursbecause their outer s electron is very easily excited.[6]:75

Caesium reacts explosively with water even at low temperatures
All the alkali metals are highly reactive and are never found in elemental forms in nature.[20] Because of this, they are usually stored inmineral oil or kerosene (paraffin oil).[21] They react aggressively with the halogens to form the alkali metal halides, which are white ionic crystalline compounds that are all soluble in water except lithium fluoride (LiF).[5] The alkali metals also react with water to form stronglyalkaline hydroxides and thus should be handled with great care. The heavier alkali metals react more vigorously than the lighter ones; for example, when dropped into water, caesium produces a larger explosion than potassium.[5][22][23] The alkali metals have the lowest firstionisation energies in their respective periods of the periodic table[24] because of their low effective nuclear charge[5] and the ability to attain a noble gas configuration by losing just one electron. The second ionisation energy of all of the alkali metals is very high[5][24] as it is in a full shell that is also closer to the nucleus;[5] thus, they almost always lose a single electron, forming cations.[6]:28 The alkalides are an exception: they are unstable compounds which contain alkali metals in a −1 oxidation state, which is very unusual as before the discovery of the alkalides, the alkali metals were not expected to be able to form anions and were thought to be able to appear in salts only as cations. The alkalide anions have filled s-subshells, which gives them more stability and allows them to exist. All the stable alkali metals except lithium are known to be able to form alkalides,[25][26][27] and the alkalides have much theoretical interest due to their unusual stoichiometry and low ionisation potentials. Alkalides are chemically similar to the electrides, which are salts with trapped electrons acting as anions.[28] A particularly striking example of an alkalide is “inverse sodium hydride“, H+Na− (both ions being complexed), as opposed to the usual sodium hydride, Na+H−:[29] it is unstable in isolation, due to its high energy resulting from the displacement of two electrons from hydrogen to sodium, although several derivatives are predicted to be metastable or stable.[29][30]
In aqueous solution, the alkali metal ions form aqua ions of the formula [M(H2O)n]+, where n is the solvation number. Their coordination numbers and shapes agree well with those expected from their ionic radii. In aqueous solution the water molecules directly attached to the metal ion are said to belong to the first coordination sphere, also known as the first, or primary, solvation shell. The bond between a water molecule and the metal ion is a dative covalent bond, with the oxygen atom donating both electrons to the bond. Each coordinated water molecule may be attached by hydrogen bonds to other water molecules. The latter are said to reside in the second coordination sphere. However, for the alkali metal cations, the second coordination sphere is not well-defined as the +1 charge on the cation is not high enough to polarize the water molecules in the primary solvation shell enough for them to form strong hydrogen bonds with those in the second coordination sphere, producing a more stable entity.[31][32]:25 The solvation number for Li+ has been experimentally determined to be 4, forming the tetrahedral [Li(H2O)4]+: while solvation numbers of 3 to 6 have been found for lithium aqua ions, solvation numbers less than 4 may be the result of the formation of contact ion-pairs, and the higher solvation numbers may be interpreted in terms of water molecules that approach [Li(H2O)4]+ through a face of the tetrahedron, though molecular dynamic simulations may indicate the existence of an octahedral hexaaqua ion. There are also probably six water molecules in the primary solvation sphere of the sodium ion, forming the octahedral [Na(H2O)6]+ ion.[7][32]:126–127 While it was previously thought that the heavier alkali metals also formed octahedral hexaaqua ions, it has since been found that potassium and rubidium probably form the [K(H2O)8]+ and [Rb(H2O)8]+ ions, which have the square antiprismaticstructure, and that caesium forms the 12-coordinate [Cs(H2O)12]+ ion.[33]
Lithium[edit]
The chemistry of lithium shows several differences from that of the rest of the group as the small Li+ cation polarises anions and gives its compounds a more covalentcharacter.[5] Lithium and magnesium have a diagonal relationship due to their similar atomic radii,[5] so that they show some similarities. For example, lithium forms a stablenitride, a property common among all the alkaline earth metals (magnesium’s group) but unique among the alkali metals.[34] In addition, among their respective groups, only lithium and magnesium form covalent organometallic compounds (e.g. LiMe and MgMe2).[35]
Lithium fluoride is the only alkali metal halide that is not soluble in water,[5] and lithium hydroxide is the only alkali metal hydroxide that is not deliquescent.[5] Conversely, lithium perchlorate and other lithium salts with large anions that cannot be polarized are much more stable than the analogous compounds of the other alkali metals, probably because Li+ has a high solvation energy.[6]:76 This effect also means that most simple lithium salts are commonly encountered in hydrated form, because the anhydrous forms are extremely hygroscopic: this allows salts like lithium chloride and lithium bromide to be used in dehumidifiers and air-conditioners.[6]:76
Francium[edit]
Francium is also predicted to show some differences due to its high atomic weight, causing its electrons to travel at considerable fractions of the speed of light and thus makingrelativistic effects more prominent. In contrast to the trend of decreasing electronegativities and ionisation energies of the alkali metals, francium’s electronegativity and ionisation energy are predicted to be higher than caesium’s due to the relativistic stabilisation of the 7s electrons; also, its atomic radius is expected to be abnormally low.[36]:1729[13][37] All known physical properties of francium also deviate from the clear trends going from lithium to caesium, such as the first ionisation energy, electron affinity, and anion polarizability.[37] The CsFr molecule is also polarized as Cs+Fr−, showing that the 7s subshell of francium is much more strongly affected by relativistic effects than the 6s subshell of caesium.[37] Additionally, francium superoxide (FrO2) is expected to have covalent character, unlike the other alkali metal superoxides, because of bonding contributions from the 6p electrons of francium.[37]
Nuclear[edit]
| Z | Alkali metal | Stable | Decays | unstable: italics
odd–odd isotopes coloured pink
|
||
|---|---|---|---|---|---|---|
| 3 | lithium | 2 | — | 7Li | 6Li | |
| 11 | sodium | 1 | — | 23Na | ||
| 19 | potassium | 2 | 1 | 39K | 41K | 40K |
| 37 | rubidium | 1 | 1 | 85Rb | 87Rb | |
| 55 | caesium | 1 | — | 133Cs | ||
| 87 | francium | — | — | No primordial isotopes (223Fr is a trace radioisotope) |
||
| Radioactive: 40K, t1/2 1.25 × 109 years; 87Rb, t1/2 4.9 × 1010 years; 223Fr, t1/2 22.0 min. | ||||||
All the alkali metals have odd atomic numbers; hence, their isotopes must be either odd–odd (both proton and neutron number are odd) or odd–even (proton number is odd, but neutron number is even). Odd–odd nuclei have even mass numbers, whereas odd–even nuclei have odd mass numbers. Odd–odd primordial nuclides are rare because most odd–odd nuclei are highly unstable with respect to beta decay, because the decay products are even–even, and are therefore more strongly bound, due to nuclear pairing effects.[38]
Due to the great rarity of odd–odd nuclei, almost all the primordial isotopes of the alkali metals are odd–even (the exceptions being the light stable isotope lithium-6 and the long-lived radioisotopepotassium-40 and rubidium-87). For a given odd mass number, there can be only a single beta-stable nuclide, since there is not a difference in binding energy between even–odd and odd–even comparable to that between even–even and odd–odd, leaving other nuclides of the same mass number (isobars) free to beta decay toward the lowest-mass nuclide. An effect of the instability of an odd number of either type of nucleons is that odd-numbered elements, such as the alkali metals, tend to have fewer stable isotopes than even-numbered elements. Of the 26 monoisotopic elements that have only a single stable isotope, all but one have an odd atomic number and all but one also have an even number of neutrons. Beryllium is the single exception to both rules, due to its low atomic number.[38]
All of the alkali metals except lithium and caesium have at least one naturally occurring radioisotope: sodium-22 and sodium-24 are trace radioisotopes producedcosmogenically,[39] potassium-40 and rubidium-87 have very long half-lives and thus occur naturally,[40] and all isotopes of francium are radioactive.[40] Caesium was also thought to be radioactive in the early 20th century,[41][42] although it has no naturally occurring radioisotopes.[40] (Francium had not been discovered yet at that time.) The natural radioisotope of potassium, potassium-40, makes up about 0.012% of natural potassium,[43] and thus natural potassium is weakly radioactive. This natural radioactivity became a basis for a mistaken claim of the discovery for element 87 (the next alkali metal after caesium) in 1925.[44][45]
Caesium-137, with a half-life of 30.17 years, is one of the two principal medium-lived fission products, along with strontium-90, which are responsible for most of the radioactivityof spent nuclear fuel after several years of cooling, up to several hundred years after use. It constitutes most of the radioactivity still left from the Chernobyl accident. 137Cs undergoes high-energy beta decay and eventually becomes stable barium-137. It is a strong emitter of gamma radiation. 137Cs has a very low rate of neutron capture and cannot be feasibly disposed of in this way, but must be allowed to decay.[46] 137Cs has been used as a tracer in hydrologic studies, analogous to the use of tritium.[47] Small amounts ofcaesium-134 and caesium-137 were released into the environment during nearly all nuclear weapon tests and some nuclear accidents, most notably the Goiânia accident and the Chernobyl disaster. As of 2005, caesium-137 is the principal source of radiation in the zone of alienation around the Chernobyl nuclear power plant.[48]
Periodic trends[edit]
The alkali metals are more similar to each other than the elements in any other group are to each other.[5] For instance, when moving down the table, all known alkali metals show increasing atomic radius,[18] decreasing electronegativity,[18] increasing reactivity,[5] and decreasing melting and boiling points[18] as well as heats of fusion and vaporisation.[6]:75 In general, their densities increase when moving down the table, with the exception that potassium is less dense than sodium.[18]
Atomic and ionic radii[edit]
Main article: Atomic radius
Effective nuclear charge on an atomic electron
The atomic radii of the alkali metals increase going down the group.[18] Because of the shielding effect, when an atom has more than one electron shell, each electron feels electric repulsion from the other electrons as well as electric attraction from the nucleus.[49] In the alkali metals, the outermost electron only feels a net charge of +1, as some of the nuclear charge (which is equal to the atomic number) is cancelled by the inner electrons; the number of inner electrons of an alkali metal is always one less than the nuclear charge. Therefore, the only factor which affects the atomic radius of the alkali metals is the number of electron shells. Since this number increases down the group, the atomic radius must also increase down the group.[18]
The ionic radii of the alkali metals are much smaller than their atomic radii. This is because the outermost electron of the alkali metals is in a different electron shell than the inner electrons, and thus when it is removed the resulting atom has one fewer electron shell and is smaller. Additionally, the effective nuclear charge has increased, and thus the electrons are attracted more strongly towards the nucleus and the ionic radius decreases.[5]
First ionisation energy[edit]
Main article: Ionisation energy

Periodic trend for ionisation energy: each period begins at a minimum for the alkali metals, and ends at a maximum for the noble gases.
The first ionisation energy of an element or molecule is the energy required to move the most loosely held electron from onemole of gaseous atoms of the element or molecules to form one mole of gaseous ions with electric charge +1. The factors affecting the first ionisation energy are the nuclear charge, the amount of shielding by the inner electrons and the distance from the most loosely held electron from the nucleus, which is always an outer electron in main group elements. The first two factors change the effective nuclear charge the most loosely held electron feels. Since the outermost electron of alkali metals always feels the same effective nuclear charge (+1), the only factor which affects the first ionisation energy is the distance from the outermost electron to the nucleus. Since this distance increases down the group, the outermost electron feels less attraction from the nucleus and thus the first ionisation energy decreases.[18] (This trend is broken in francium due to therelativistic stabilization and contraction of the 7s orbital, bringing francium’s valence electron closer to the nucleus than would be expected from non-relativistic calculations. This makes francium’s outermost electron feel more attraction from the nucleus, increasing its first ionisation energy slightly beyond that of caesium.)[36]:1729
The second ionisation energy of the alkali metals is much higher than the first as the second-most loosely held electron is part of a fully filled electron shell and is thus difficult to remove.[5]
Reactivity[edit]
Main article: Reactivity (chemistry)
The reactivities of the alkali metals increase going down the group. This is the result of a combination of two factors: the first ionisation energies and atomisation energies of the alkali metals. Because the first ionisation energy of the alkali metals decreases down the group, it is easier for the outermost electron to be removed from the atom and participate in chemical reactions, thus increasing reactivity down the group. The atomisation energy measures the strength of the metallic bond of an element, which falls down the group as the atoms increase in radius and thus the metallic bond must increase in length, making the delocalised electrons further away from the attraction of the nuclei of the heavier alkali metals. Adding the atomisation and first ionisation energies gives a quantity closely related to (but not equal to) the activation energy of the reaction of an alkali metal with another substance. This quantity decreases going down the group, and so does the activation energy; thus, chemical reactions can occur faster and the reactivity increases down the group.[50]
Electronegativity[edit]
Main article: Electronegativity

The variation of Pauling electronegativity (y-axis) as one descends the main groups of the periodic table from the second to the sixth period
Electronegativity is a chemical property that describes the tendency of an atom or a functional group to attract electrons (orelectron density) towards itself.[51] If the bond between sodium and chlorine in sodium chloride were covalent, the pair of shared electrons would be attracted to the chlorine because the effective nuclear charge on the outer electrons is +7 in chlorine but is only +1 in sodium. The electron pair is attracted so close to the chlorine atom that they are practically transferred to the chlorine atom (an ionic bond). However, if the sodium atom was replaced by a lithium atom, the electrons will not be attracted as close to the chlorine atom as before because the lithium atom is smaller, making the electron pair more strongly attracted to the closer effective nuclear charge from lithium. Hence, the larger alkali metal atoms (further down the group) will be less electronegative as the bonding pair is less strongly attracted towards them.[18]
Because of the higher electronegativity of lithium, some of its compounds have a more covalent character. For example,lithium iodide (LiI) will dissolve in organic solvents, a property of most covalent compounds.[18] Lithium fluoride (LiF) is the only alkali halide that is not soluble in water,[5] and lithium hydroxide (LiOH) is the only alkali metal hydroxide that is notdeliquescent.[5]
Melting and boiling points[edit]
Main articles: Melting point and Boiling point
The melting point of a substance is the point where it changes state from solid to liquid while the boiling point of a substance (in liquid state) is the point where the vapour pressure of the liquid equals the environmental pressure surrounding the liquid[52][53] and all the liquid changes state to gas. As a metal is heated to its melting point, the metallic bonds keeping the atoms in place weaken so that the atoms can move around, and the metallic bonds eventually break completely at the metal’s boiling point.[18][54] Therefore, the falling melting and boiling points of the alkali metals indicate that the strength of the metallic bonds of the alkali metals decreases down the group.[18] This is because metal atoms are held together by the electromagnetic attraction from the positive ions to the delocalised electrons.[18][54] As the atoms increase in size going down the group (because their atomic radius increases), the nuclei of the ions move further away from the delocalised electrons and hence the metallic bond becomes weaker so that the metal can more easily melt and boil, thus lowering the melting and boiling points.[18] (The increased nuclear charge is not a relevant factor due to the shielding effect.)[18]
Density[edit]
Main article: Density
The alkali metals all have the same crystal structure (body-centred cubic)[6] and thus the only relevant factors are the number of atoms that can fit into a certain volume and the mass of one of the atoms, since density is defined as mass per unit volume. The first factor depends on the volume of the atom and thus the atomic radius, which increases going down the group; thus, the volume of an alkali metal atom increases going down the group. The mass of an alkali metal atom also increases going down the group. Thus, the trend for the densities of the alkali metals depends on their atomic weights and atomic radii; if figures for these two factors are known, the ratios between the densities of the alkali metals can then be calculated. The resultant trend is that the densities of the alkali metals increase down the table, with an exception at potassium. Due to having the lowest atomic weight of all the elements in their period and having the largest atomic radius for their periods, the alkali metals are the least dense metals in the periodic table.[18] Lithium, sodium, and potassium are the only three metals in the periodic table that are less dense than water:[5] in fact, lithium is the least dense known solid at room temperature.[6]:75
Compounds[edit]
Hydroxides[edit]

A reaction of 3 pounds (≈ 1.4 kg) of sodium with water
All the alkali metals react vigorously or explosively with cold water, producing an aqueous solution of a strongly basic alkali metalhydroxide and releasing hydrogen gas.[50] This reaction becomes more vigorous going down the group: lithium reacts steadily witheffervescence, but sodium and potassium can ignite and rubidium and caesium sink in water and generate hydrogen gas so rapidly that shock waves form in the water that may shatter glass containers.[5] When an alkali metal is dropped into water, it produces an explosion, of which there are two separate stages. The metal reacts with the water first, breaking the hydrogen bonds in the water and producing hydrogen gas; this takes place faster for the more reactive heavier alkali metals. Second, the heat generated by the first part of the reaction often ignites the hydrogen gas, causing it to burn explosively into the surrounding air. This secondary hydrogen gas explosion produces the visible flame above the bowl of water, lake or other body of water, not the initial reaction of the metal with water (which tends to happen mostly under water).[22] The alkali metal hydroxides are the most basic known hydroxides.[6]:87
Recent research has suggested that the explosive behavior of alkali metals in water is driven by a Coulomb explosion rather than solely by rapid generation of hydrogen itself.[55] All alkali metals melt as a part of the reaction with water. Water molecules ionize the bare metallic surface of the liquid metal, leaving a positively charged metal surface and negatively charged water ions. The attraction between the charged metal and water ions will rapidly increase the surface area, causing an exponential increase of ionization. When the repulsive forces within the liquid metal surface exceeds the forces of the surface tension, it vigorously explodes.
Compounds with the group 14 elements[edit]


Lithium and sodium react with carbon to form acetylides, Li2C2 and Na2C2, which can also be obtained by reaction of the metal with acetylene. Potassium, rubidium, and caesium react with graphite; their atoms are intercalated between the hexagonal graphite layers, forming graphite intercalation compounds of formulae MC60 (dark grey, almost black), MC48(dark grey, almost black), MC36 (blue), MC24 (steel blue), and MC8 (bronze) (M = K, Rb, or Cs). These compounds are over 200 times more electrically conductive than pure graphite, suggesting that the valence electron of the alkali metal is transferred to the graphite layers (e.g. M+
C−
8).[7] Upon heating of KC8, the elimination of potassium atoms results in the conversion in sequence to KC24, KC36, KC48 and finally KC60. KC8 is a very strong reducing agent and is pyrophoric and explodes on contact with water.[56][57] While the large alkali metals (K, Rb, and Cs) initially form MC8, the smaller ones initially form MC6.[58]
When the alkali metals react with the heavier elements in the carbon group, ionic substances with cage-like structures are formed, such as the silicide M4Si4 (M = K, Rb, or Cs), which contains M+ and tetrahedral Si4−
4 ions.[7] The chemistry of alkali metal germanides, involving the germanide ion Ge4− and other cluster (Zintl) ions such as Ge2−
4, Ge4−
9, Ge2−
9, and [(Ge9)2]6−, is largely analogous to that of the corresponding silicides.[6] Alkali metal stannides are mostly ionic, sometimes with the stannide ion (Sn4−),[59] and sometimes with more complex Zintl ions such as Sn4−
9, which appears in tetrapotassium nonastannide (K4Sn9).[60] The monatomic plumbide ion (Pb4−) is unknown, and indeed its formation is predicted to be energetically unfavourable; alkali metal plumbides have complex Zintl ions, such as Pb4−
9.[6]
Nitrides and pnictides[edit]

Unit cell ball-and-stick model oflithium nitride[61] On the basis of size atetrahedral structure would be expected, but that would be geometrically impossible: thus lithium nitride takes on this unique crystal structure.[6]:76
Lithium, the lightest of the alkali metals, is the only alkali metal which reacts with nitrogen at standard conditions, and its nitride is the only stable alkali metal nitride. Nitrogen is an unreactive gas because breaking the strong triple bond in the dinitrogen molecule (N2) requires a lot of energy. The formation of an alkali metal nitride would consume the ionisation energy of the alkali metal (forming M+ ions), the energy required to break the triple bond in N2 and the formation of N3− ions, and all the energy released from the formation of an alkali metal nitride is from the lattice energy of the alkali metal nitride. The lattice energy is maximised with small, highly charged ions; the alkali metals do not form highly charged ions, only forming ions with a charge of +1, so only lithium, the smallest alkali metal, can release enough lattice energy to make the reaction with nitrogen exothermic, forming lithium nitride. The reactions of the other alkali metals with nitrogen would not release enough lattice energy and would thus be endothermic, so they do not form nitrides at standard conditions.[34](Sodium nitride (Na3N) and potassium nitride (K3N), while existing, are extremely unstable, being prone to decomposing back into their constituent elements, and cannot be produced by reacting the elements with each other at standard conditions.)[62][63]
All the alkali metals react readily with phosphorus and arsenic to form phosphides and arsenides with the formula M3Pn (where M represents an alkali metal and Pn represents a pnictogen). This is due to the greater size of the P3− and As3− ions, so that less lattice energy needs to be released for the salts to form.[7] These are not the only phosphides and arsenides of the alkali metals: for example, potassium has nine different known phosphides, with formulae K3P, K4P3, K5P4, KP, K4P6, K3P7, K3P11, KP10.3, and KP15.[64] While most metals form arsenides, only the alkali and alkaline earth metals form mostly ionic arsenides. The structure of Na3As is complex with unusually short Na–Na distances of 328–330 pm which are shorter than in sodium metal, and this indicates that even with these electropositive metals the bonding cannot be straightforwardly ionic.[6] Other alkali metal arsenides not conforming to the formula M3As are known, such as LiAs, which has a metallic lustre and electrical conductivity indicating the presence of some metallic bonding.[6] The antimonides are unstable and reactive as the Sb3− ion is a strong reducing agent; reaction of them with acids form the toxic and unstable gas stibine (SbH3).[65] Bismuthides are not even wholly ionic; they are intermetallic compounds containing partially metallic and partially ionic bonds.[66]
Oxides and chalcogenides[edit]
See also: Alkali metal oxide

Rb
9O
2 cluster, composed of two regular octahedraconnected to each other by one face
9O
2 cluster, composed of two regular octahedraconnected to each other by one face

Cs
11O
3 cluster, composed of three regular octahedra where each octahedron is connected to both of the others by one face each. All three octahedra have one edge in common.
11O
3 cluster, composed of three regular octahedra where each octahedron is connected to both of the others by one face each. All three octahedra have one edge in common.
All the alkali metals react vigorously with oxygen at standard conditions. They form various types of oxides, such as simpleoxides (containing the O2− ion), peroxides (containing the O2−
2 ion, where there is a single bond between the two oxygen atoms), superoxides (containing the O−
2 ion), and many others. Lithium burns in air to form lithium oxide, but sodium reacts with oxygen to form a mixture of sodium oxide and sodium peroxide. Potassium forms a mixture of potassium peroxide andpotassium superoxide, while rubidium and caesium form the superoxide exclusively. Their reactivity increases going down the group: while lithium, sodium and potassium merely burn in air, rubidium and caesium are pyrophoric (spontaneously catch fire in air).[34]
The smaller alkali metals tend to polarise the more complex anions (the peroxide and superoxide) due to their small size. This attracts the electrons in the more complex anions towards one of its constituent oxygen atoms, forming an oxide ion and an oxygen atom. This causes lithium to form the oxide exclusively on reaction with oxygen at room temperature. This effect becomes drastically weaker for the larger sodium and potassium, allowing them to form the less stable peroxides. Rubidium and caesium, at the bottom of the group, are so large that even the least stable superoxides can form. Because the superoxide releases the most energy when formed, the superoxide is preferentially formed for the larger alkali metals where the more complex anions are not polarised. (The oxides and peroxides for these alkali metals do exist, but do not form upon direct reaction of the metal with oxygen at standard conditions.)[34] In addition, the small size of the Li+ and O2− ions contributes to their forming a stable ionic lattice structure. Under controlled conditions, however, all the alkali metals, with the exception of francium, are known to form their oxides, peroxides, and superoxides. The alkali metal peroxides and superoxides are powerful oxidizing agents. Sodium peroxide and potassium superoxide react with carbon dioxide to form the alkali metal carbonate and oxygen gas, which allows them to be used in submarine air purifiers; the presence of water vapour, naturally present in breath, makes the removal of carbon dioxide by potassium superoxide even more efficient.[7][67] All the stable alkali metals except lithium can form red ozonides (MO3) through low-temperature reaction of the powdered anhydrous hydroxide with ozone: the ozonides may be then extracted using liquid ammonia.[6]:85
Rubidium and caesium can form even more complicated oxides than the superoxides. Rubidium can form Rb6O and Rb9O2 upon oxidation in air, while caesium forms an immense variety of oxides, such as the ozonide CsO3[68][69] and several brightly coloured suboxides,[70] such as Cs
7O, Cs
4O, Cs
11O
3, Cs
3O (dark-green[71]), CsO, Cs
3O
2,[72] as well as Cs
7O
2.[73][74] The latter may be heated under vacuum to generate Cs
2O.[75]
The alkali metals can also react analogously with the heavier chalcogens (sulfur, selenium, tellurium, and polonium), and all the alkali metal chalcogenides are known (with the exception of francium’s). Reaction with an excess of the chalcogen can similarly result in lower chalcogenides, with chalcogen ions containing chains of the chalcogen atoms in question. For example, sodium can react with sulfur to form the sulfide (Na2S) and various polysulfides with the formula Na2Sx (x from 2 to 6), containing the S2−
x ions.[7] Due to the basicity of the Se2− and Te2− ions, the alkali metal selenides and tellurides are alkaline in solution; when reacted directly with selenium and tellurium, alkali metal polyselenides and polytellurides are formed along with the selenides and tellurides with the Se2−
x and Te2−
x ions.[76] The alkali metal polonides are all ionic compounds containing the Po2− ion; they are very chemically stable and can be produced by direct reaction of the elements at around 300–400 °C.[6][77][78]
Hydrides and halides[edit]
Main article: Alkali metal halide
The alkali metals are among the most electropositive elements on the periodic table and thus tend to bond ionically to the most electronegative elements on the periodic table, the halogens, forming salts known as the alkali metal halides. The reaction is very vigorous and can sometimes result in explosions.[6]:76 This includes sodium chloride, otherwise known as common salt. The reactivity becomes higher from lithium to caesium and drops from fluorine to iodine. All of the alkali metal halides have the formula MX where M is an alkali metal and X is a halogen. They are all white ionic crystalline solids.[5][34] All the alkali metal halides are soluble in water except for lithium fluoride (LiF), which is insoluble in water due to its very high lattice enthalpy. The high lattice enthalpy of lithium fluoride is due to the small sizes of the Li+ and F− ions, causing the electrostatic interactions between them to be strong:[5] a similar effect occurs for magnesium fluoride, which lithium has a diagonal relationship with.[6]:76 The alkali metals also react similarly with hydrogen to form ionic alkali metal hydrides.[7]
Coordination complexes[edit]

18-crown-6 coordinating a potassium ion

Structure of 2.2.2-Cryptandencapsulating a potassium cation (purple). At crystalline state, obtained with an X-ray diffraction.[79]
Alkali metal cations do not usually form coordination complexes with simple Lewis bases due to their low charge of just +1 and their relatively large size; thus the Li+ ion forms most complexes and the heavier alkali metal ions form less and less. Inaqueous solution, the alkali metal ions exist as octahedral hexahydrate complexes ([M(H2O)6)]+), with the exception of the lithium ion, which due to its small size forms tetrahedral tetrahydrate complexes ([Li(H2O)4)]+); the alkali metals form these complexes because their ions are attracted by electrostatic forces of attraction to the polar water molecules. Because of this,anhydrous salts containing alkali metal cations are often used as desiccants.[7] Alkali metals also readily form complexes withcrown ethers (e.g. 12-crown-4 for Li+, 15-crown-5 for Na+, and 18-crown-6 for K+) and cryptands due to electrostatic attraction.[7]
Ammonia solutions[edit]
The alkali metals dissolve slowly in liquid ammonia, forming hydrogen gas and the alkali metal amide (MNH2, where M represents an alkali metal): this was first noted by Humphry Davy in 1809 and rediscovered by W. Weyl in 1864. The process may be speeded up by a catalyst. Similar solutions are formed by the heavy divalent alkaline earth metals and lanthanides calcium, strontium, barium, europium, andytterbium. The amide salt is quite insoluble and readily precipitates out of solution, leaving intensely coloured ammonia solutions of the alkali metals. In 1907, Charles Krause identified the colour as being due to the presence of solvated electrons, which contribute to the high electrical conductivity of these solutions. At low concentrations (below 3 M), the solution is dark blue and has ten times the conductivity of aqueous sodium chloride; at higher concentrations (above 3 M), the solution is copper-coloured and has approximately the conductivity of liquid metals like mercury.[6][7][80] In addition to the alkali metal amide salt and solvated electrons, such ammonia solutions also contain the alkali metal cation (M+), the neutral alkali metal atom (M), diatomic alkali metal molecules (M2) and alkali metal anions (M−). These are unstable and eventually become the more thermodynamically stable alkali metal amide and hydrogen gas. Solvated electrons are powerful reducing agents and are often used in chemical synthesis.[7]
Organometallic[edit]

Structure of the methyllithium tetramer, (CH3Li)4
Being the smallest alkali metal, lithium forms the widest variety of and most stable organometallic compounds, which are bonded covalently.Organolithium compounds are electrically non-conducting volatile solids or liquids that melt at low temperatures, and tend to form oligomerswith the structure (RLi)x where R is the organic group. As the electropositive nature of lithium puts most of the charge density of the bond on the carbon atom, effectively creating a carbanion, organolithium compounds are extremely powerful bases and nucleophiles. For use as bases, butyllithiums are often used and are commercially available. An example of an organolithium compound is methyllithium ((CH3Li)x), which exists in tetrameric (x = 4) and hexameric (x = 6) forms.[7][81]
The application of organosodium compounds in chemistry is limited in part due to competition from organolithium compounds, which are commercially available and exhibit more convenient reactivity. The principal organosodium compound of commercial importance is sodium cyclopentadienide. Sodium tetraphenylborate can also be classified as an organosodium compound since in the solid state sodium is bound to the aryl groups. Organometallic compounds of the higher alkali metals are even more reactive than organosodium compounds and of limited utility. A notable reagent is Schlosser’s base, a mixture of n-butyllithium and potassium tert-butoxide. This reagent reacts with propeneto form the compound allylpotassium (KCH2CHCH2). cis-2-Butene and trans-2-butene equilibrate when in contact with alkali metals. Whereasisomerization is fast with lithium and sodium, it is slow with the higher alkali metals. The higher alkali metals also favour the sterically congested conformation.[82] Several crystal structures of organopotassium compounds have been reported, establishing that they, like the sodium compounds, are polymeric.[83] Organosodium, organopotassium, organorubidium and organocaesium compounds are all mostly ionic and are insoluble (or nearly so) in nonpolar solvents.[7]
Extensions[edit]
See also: Ununennium

Although francium is the heaviest alkali metal that has been discovered, there has been some theoretical work predicting the physical and chemical characteristics of the hypothetical heavier alkali metals. Being the first period 8 element, the undiscovered elementununennium (element 119) is predicted to be the next alkali metal after francium and behave much like their lighter congeners; however, it is also predicted to differ from the lighter alkali metals in some properties.[36]:1729–1730 Its chemistry is predicted to be closer to that of potassium[85] or rubidium[36]:1729–1730 instead of caesium or francium. This is unusual as periodic trends, ignoring relativistic effects would predict ununennium to be even more reactive than caesium and francium. This lowered reactivity is due to the relativistic stabilisation of ununennium’s valence electron, increasing ununennium’s first ionisation energy and decreasing themetallic and ionic radii;[85] this effect is already seen for francium.[36]:1729–1730 This assumes that ununennium will behave chemically as an alkali metal, which, although likely, may not be true due to relativistic effects.[86] The relativistic stabilisation of the 8s orbital also increases ununennium’s electron affinity far beyond that of caesium and francium; indeed, ununennium is expected to have an electron affinity higher than all the alkali metals lighter than it. Relativistic effects also cause a very large drop in the polarisability of ununennium.[36]:1729–1730 On the other hand, ununennium is predicted to continue the trend of melting points decreasing going down the group, being expected to have a melting point between 0 °C and 30 °C.[36]:1724

Empirical (Na–Fr) and predicted (Uue) electron affinity of the alkali metals from the third to the eighth period, measured in electron volts[36]:1730[84]
The stabilisation of ununennium’s valence electron and thus the contraction of the 8s orbital cause its atomic radius to be lowered to 240 pm,[36]:1729–1730 very close to that of rubidium (247 pm),[5] so that the chemistry of ununennium in the +1 oxidation state should be more similar to the chemistry of rubidium than to that of francium. On the other hand, the ionic radius of the Uue+ ion is predicted to be larger than that of Rb+, because the 7p orbitals are destabilised and are thus larger than the p-orbitals of the lower shells. Ununennium may also show the +3oxidation state,[36]:1729–1730 which is not seen in any other alkali metal,[6]:28 in addition to the +1 oxidation state that is characteristic of the other alkali metals and is also the main oxidation state of all the known alkali metals: this is because of the destabilisation and expansion of the 7p3/2 spinor, causing its outermost electrons to have a lower ionisation energy than what would otherwise be expected.[6]:28[36]:1729–1730Indeed, many ununennium compounds are expected to have a large covalent character, due to the involvement of the 7p3/2 electrons in the bonding.[37]

Not as much work has been done predicting the properties of the alkali metals beyond ununennium. Although a simple extrapolation of the periodic table would put element 169, unhexennium, under ununennium, Dirac-Fock calculations predict that the next alkali metal after ununennium may actually be element 165, unhexpentium, which is predicted to have the electron configuration [Uuo] 5g18 6f14 7d10 8s2 8p1/22 9s1.[36]:1729–1730[84] Further calculations show that unhexpentium would follow the trend of increasing ionisation energy beyond caesium, having an ionisation energy comparable to that of sodium, and that it should also continue the trend of decreasing atomic radii beyond caesium, having an atomic radius comparable to that of potassium.[36]:1729–1730 However, the 7d electrons of unhexpentium may also be able to participate in chemical reactions along with the 9s electron, possibly allowing oxidation states beyond +1 and perhaps even making unhexpentium behave more like a boron group element orgroup 11 element than an alkali metal.[36]:1732–1733[87] Due to the alkali and alkaline earth metals both being s-block elements, these predictions for the trends and properties of ununennium and unhexpentium also mostly hold quite similarly for the corresponding alkaline earth metals unbinilium (Ubn) and unhexhexium (Uhh).[36]:1729–1733
The probable properties of further alkali metals beyond unhexpentium have not been explored yet as of 2012. In periods 8 and above of the periodic table, relativistic and shell-structure effects become so strong that extrapolations from lighter congeners become completely inaccurate. In addition, the relativistic and shell-structure effects (which stabilise the s-orbitals and destabilise and expand the d-, f-, and g-orbitals of higher shells) have opposite effects, causing even larger difference between relativistic and non-relativistic calculations of the properties of elements with such high atomic numbers.[36]:1732–1733 Interest in the chemical properties of ununennium and unhexpentium stems from the fact that both elements are located close to the expected locations of islands of stabilities, centered at elements 122 (306Ubb) and 164 (482Uhq).[88][89][90]
Other similar substances[edit]
Hydrogen[edit]
Main article: Hydrogen

Hydrogen gas glowing in adischarge tube
The element hydrogen, with one electron per neutral atom, is usually placed at the top of Group 1 of the periodic table for convenience, but hydrogen is not normally considered to be an alkali metal;[91] when it is considered to be an alkali metal, it is because of its atomic properties and not its chemical properties.[92] Under typical conditions, pure hydrogen exists as a diatomic gas consisting of two atoms permolecule (H2);[93] however, the alkali metals only form diatomic molecules (such as dilithium, Li2) at high temperatures, when they are in the gaseous state.[94]
Hydrogen, like the alkali metals, has one valence electron[95] and reacts easily with the halogens[95] but the similarities end there.[95] Its placement above lithium is primarily due to its electron configuration and not its chemical properties.[91][95] It is sometimes placed abovecarbon due to their similar electronegativities[96] or fluorine due to their similar chemical properties.[95][96]
The first ionisation energy of hydrogen (1312.0 kJ/mol) is much higher than that of the alkali metals.[97][98] As only one additional electron is required to fill in the outermost shell of the hydrogen atom, hydrogen often behaves like a halogen, forming the negative hydride ion, and is sometimes considered to be a halogen.[95] (The alkali metals can also form negative ions, known as alkalides, but these are little more than laboratory curiosities, being unstable.)[29][30] It was expected for some time that liquid hydrogen would show metallic properties;[96] while this has been shown to not be the case, under extremely high pressures, such as those found at the cores of Jupiter and Saturn, hydrogen does become metallic and behaves like an alkali metal; in this phase, it is known as metallic hydrogen.[99] The electrical resistivity of liquid metallic hydrogen at 3000 K is approximately equal to that of liquid rubidium and caesium at 2000 K at the respective pressures when they undergo a nonmetal-to-metal transition.[100]
The 1s1 electron configuration of hydrogen, while superficially similar to that of the alkali metals (ns1), is unique because there is no 1p subshell. Hence it can lose an electron to form the hydron H+, or gain one to form the hydride ion H−.[6]:43 In the former case it resembles superficially the alkali metals; in the latter case, the halogens, but the differences due to the lack of a 1p subshell are important enough that neither group fits the properties of hydrogen well.[6]:43 Group 14 is the best fit in terms of thermodynamic properties such as ionization energy and electron affinity, but none of the three placements are entirely satisfactory.[96] As an example of hydrogen’s unorthodox properties stemming from its unusual electron configuration and small size, the hydrogen ion is very small (radius around 150 fm compared to the 50–220 pm size of most other atoms and ions) and so is nonexistent in condensed systems other than in association with other atoms or molecules. Indeed, transferring of protons between chemicals is the basis of acid-base chemistry.[6]:43 Also unique is hydrogen’s ability to form hydrogen bonds, which are an effect of charge-transfer, electrostatic, and electron correlative contributing phenomena.[96]While analogous lithium bonds are also known, they are mostly electrostatic.[96] Nevertheless, hydrogen can perform the same structural role as the alkali metals in some molecular crystals, and has a close relationship with the lightest alkali metals (especially lithium).[101]
Ammonium[edit]
Main article: Ammonium
The ammonium ion (NH+
4) has very similar properties to the heavier alkali metals, acting as an alkali metal intermediate between potassium and rubidium,[102] and is often considered a close relative.[103][104][105] For example, most alkali metal salts are soluble in water, a property which ammonium salts share.[106] Ammonium is expected to behave stably as a metal (NH+
4 ions in a sea of electrons) at very high pressures (though less than the typical pressure where transitions from insulating to metallic behaviour occur around, 100 GPa), and could possibly occur inside the ice giants Uranus and Neptune, which may have significant impacts on their interior magnetic fields.[104][105] It has been estimated that the transition from a mixture of ammonia and dihydrogen molecules to metallic ammonium may occur at pressures just below 25 GPa.[104]
Thallium[edit]
Main article: Thallium
Thallium displays the +1 oxidation state[6]:28 that all the known alkali metals display,[6]:28 and thallium compounds with thallium in its +1oxidation state closely resemble the corresponding potassium or silver compounds stoichiometrically due to the similar ionic radii of the Tl+(164 pm), K+ (152 pm) and Ag+ (129 pm) ions.[107][108] It was sometimes considered an alkali metal in continental Europe (but not in England) in the years immediately following its discovery,[108]:126 and was placed just after caesium as the sixth alkali metal in Dmitri Mendeleev‘s 1869 periodic table and Julius Lothar Meyer‘s 1868 periodic table.[109] (Mendeleev’s 1871 periodic table and Meyer’s 1870 periodic table put thallium in its current position in the boron group and leave the space below caesium blank.)[109] However, thallium also displays the oxidation state +3,[6]:28 which no known alkali metal displays[6]:28 (although ununennium, the undiscovered seventh alkali metal, is predicted to possibly display the +3 oxidation state).[36]:1729–1730 The sixth alkali metal is now considered to be francium.[110]While Tl+ is stabilized by the inert pair effect, this inert pair of 6s electrons is still able to participate chemically, so that these electrons are stereochemically active in aqueous solution. Additionally, the thallium halides (except TlF) are quite insoluble in water, and TlI has an unusual structure because of the presence of the inert pair in thallium.[111]
Copper, silver, and gold[edit]
Copper
Silver
Gold
The group 11 metals (or coinage metals), copper, silver, and gold, are typically categorised as transition metals given they can form ions with incomplete d-shells. Physically, they have the relatively low melting points and high electronegativity values associated with post-transition metals. “The filled d subshell and free s electron of Cu, Ag, and Au contribute to their high electrical and thermal conductivity. Transition metals to the left of group 11 experience interactions between s electrons and the partially filled d subshell that lower electron mobility.”[112] Chemically, the group 11 metals behave like main-group metals in their +1 valence states, and are hence somewhat related to the alkali metals: this is one reason for their previously being labelled as “group IB”, paralleling the alkali metals’ “group IA”. They are occasionally classified as post-transition metals.[113] Their spectra are however analogous to those of the alkali metals.[114]
In Mendeleev’s 1871 periodic table, copper, silver, and gold are listed twice, once under group VIII (with the iron triad and platinum group metals), and once under group IB. Group IB was nonetheless parenthesized to note that it was tentative. Mendeleev’s main criterion for group assignment was the maximum oxidation state of an element: on that basis, the group 11 elements could not be classified in group IB, due to the existence of Cu(II) and Au(III) compounds being known at that time.[114] However, eliminating group IB would make group I the only main group (group VIII was labelled a transition group) to lack an A–B bifurcation.[114] Soon afterwards, a majority of chemists chose to classify these elements in group IB and remove them from group VIII for the resulting symmetry: this was the predominant classification until the rise of the modern medium-long 18-column periodic table, which separated the alkali metals and group 11 metals.[114]
The coinage metals were traditionally regarded as a subdivision of the alkali metal group, due to them sharing the characteristic s1 electron configuration of the alkali metals (group 1: p6s1; group 11: d10s1). However, the similarities are largely confined to the stochiometries of the +1 compounds of both groups, and not their chemical properties.[6]:1177This stems from the filled d subshell providing a much weaker shielding effect on the outermost s electron than the filled p subshell, so that the coinage metals have much higher first ionization energies and smaller ionic radii than do the corresponding alkali metals.[6]:1177 Furthermore, they have higher melting points, hardnesses, and densities, and lower reactivities and solubilities in liquid ammonia, as well as having more covalent character in their compounds.[6]:1177 Finally, the alkali metals are at the top of the electrochemical series, whereas the coinage metals are almost at the very bottom.[6]:1177 The coinage metals’ filled d shell is much more easily disrupted than the alkali metals’ filled p shell, so that the second and third ionization energies are lower, enabling higher oxidation states than +1 and a richer coordination chemistry, thus giving the group 11 metals cleartransition metal character.[6]:1177 Particularly noteworthy is gold forming ionic compounds with rubidium and caesium, in which it forms the auride ion (Au−) which also occurs in solvated form in liquid ammonia solution: here gold behaves as a pseudohalogen because its 5d106s1 configuration has one electron less than the quasi-closed shell 5d106s2configuration of mercury.[6]:1177
History[edit]

Petalite, the lithium mineral from which lithium was first isolated
Sodium compounds have been known since ancient times; salt (sodium chloride) has been an important commodity in human activities, as testified by the English word salary, referring to salarium, money paid to Roman soldiers for the purchase of salt.[115] While potash has been used since ancient times, it was not understood for most of its history to be a fundamentally different substance from sodium mineral salts. Georg Ernst Stahl obtained experimental evidence which led him to suggest the fundamental difference of sodium and potassium salts in 1702,[116] and Henri Louis Duhamel du Monceau was able to prove this difference in 1736.[117] The exact chemical composition of potassium and sodium compounds, and the status as chemical element of potassium and sodium, was not known then, and thus Antoine Lavoisier did include the alkali in his list of chemical elements in 1789.[118][119]
Pure potassium was first isolated in 1807 in England by Sir Humphry Davy, who derived it from caustic potash (KOH, potassium hydroxide) by the use of electrolysis of the molten salt with the newly invented voltaic pile. Previous attempts at electrolysis of the aqueous salt were unsuccessful due to potassium’s extreme reactivity.[6]:68 Potassium was the first metal that was isolated by electrolysis.[120] Later that same year, Davy reported extraction of sodium from the similar substance caustic soda (NaOH, lye) by a similar technique, demonstrating the elements, and thus the salts, to be different.[118][119][121][122] Later that year, the first pieces of pure molten sodium metal were similarly prepared by Humphry Davy through the electrolysis of molten caustic soda (now called sodium hydroxide).[121]

Johann Wolfgang Döbereiner was among the first to notice similarities between what are now known as the alkali metals.
Petalite (LiAlSi4O10) was discovered in 1800 by the Brazilian chemist José Bonifácio de Andrada in a mine on the island of Utö, Sweden.[123][124][125]However, it was not until 1817 that Johan August Arfwedson, then working in the laboratory of the chemist Jöns Jacob Berzelius, detected the presence of a new element while analysing petalite ore.[126][127] This new element was noted by him to form compounds similar to those of sodium and potassium, though its carbonate and hydroxide were less soluble in water and more alkaline than the other alkali metals.[128] Berzelius gave the unknown material the name “lithion/lithina“, from the Greek word λιθoς (transliterated as lithos, meaning “stone”), to reflect its discovery in a solid mineral, as opposed to potassium, which had been discovered in plant ashes, and sodium, which was known partly for its high abundance in animal blood. He named the metal inside the material “lithium“.[20][124][127] Lithium, sodium, and potassium were part of the discovery of periodicity, as they are among a series of triads of elements in the same group that were noted by Johann Wolfgang Döbereiner in 1850 as having similar properties.[109]

Lepidolite, the rubidium mineral from which rubidium was first isolated
Rubidium and caesium were the first elements to be discovered using the spectroscope, invented in 1859 by Robert Bunsen and Gustav Kirchhoff.[129] The next year, they discovered caesium in the mineral water from Bad Dürkheim, Germany. Their discovery of rubidium came the following year in Heidelberg, Germany, finding it in the mineral lepidolite.[130] The names of rubidium and caesium come from the most prominent lines in their emission spectra: a bright red line for rubidium (from the Latin word rubidus, meaning dark red or bright red), and a sky-blue line for caesium (derived from the Latin word caesius, meaning sky-blue).[131][note 10][132]
Around 1865 John Newlands produced a series of papers where he listed the elements in order of increasing atomic weight and similar physical and chemical properties that recurred at intervals of eight; he likened such periodicity to the octaves of music.[133][134] His version put all the alkali metals then known (lithium to caesium), as well as copper, silver, and thallium (which show the +1 oxidation state characteristic of the alkali metals), together into a group. His table placed hydrogen with the halogens.[109]

Mendeleev’s periodic system proposed in 1871 showing hydrogen and the alkali metals as part of his group I
After 1869, Dmitri Mendeleev proposed his periodic table placing lithium at the top of a group with sodium, potassium, rubidium, caesium, and thallium.[135] Two years later, Mendeleev revised his table, placing hydrogen in group 1 above lithium, and also moving thallium to the boron group. In this 1871 version, copper, silver, and gold were placed twice, once as part of group IB, and once as part of a “group VIII” encompassing today’s groups 8 to 11.[114][note 11] After the introduction of the 18-column table, the group IB elements were moved to their current position in thed-block, while alkali metals were left in group IA. Later the group’s name was changed to group 1 in 1988.[136] The trivial name “alkali metals” comes from the fact that the hydroxides of the group 1 elements are all strong alkalis when dissolved in water.[5]
There were at least four erroneous and incomplete discoveries[44][45][137][138] beforeMarguerite Perey of the Curie Institute in Paris, France discovered francium in 1939 by purifying a sample of actinium-227, which had been reported to have a decay energy of 220 keV. However, Perey noticed decay particles with an energy level below 80 keV. Perey thought this decay activity might have been caused by a previously unidentified decay product, one that was separated during purification, but emerged again out of the pureactinium-227. Various tests eliminated the possibility of the unknown element being thorium,radium, lead, bismuth, or thallium. The new product exhibited chemical properties of an alkali metal (such as coprecipitating with caesium salts), which led Perey to believe that it was element 87, caused by the alpha decay of actinium-227.[139] Perey then attempted to determine the proportion of beta decay to alpha decay in actinium-227. Her first test put the alpha branching at 0.6%, a figure that she later revised to 1%.[140] It was the last element discovered in nature, rather than by synthesis.[note 12]
The next element below francium (eka-francium) is very likely to be ununennium (Uue), element 119,[36]:1729–1730 although this is not completely certain due to relativistic effects.[86]The synthesis of ununennium was first attempted in 1985 by bombarding a target ofeinsteinium-254 with calcium-48 ions at the superHILAC accelerator at Berkeley, California. No atoms were identified, leading to a limiting yield of 300 nb.[141][142]
It is highly unlikely[141] that this reaction will be able to create any atoms of ununennium in the near future, given the extremely difficult task of making sufficient amounts of 254Es, which is favoured for production of ultraheavy elements because of its large mass, relatively long half-life of 270 days, and availability in significant amounts of several micrograms,[143] to make a large enough target to increase the sensitivity of the experiment to the required level; einsteinium has not been found in nature and has only been produced in laboratories. However, given that ununennium is only the first period 8 element on the extended periodic table, it may well be discovered in the near future through other reactions; indeed, another attempt to synthesise ununennium by bombarding a berkelium target with titanium ions is under way at the GSI Helmholtz Centre for Heavy Ion Research in Darmstadt, Germany.[144] Currently, none of the period 8 elements have been discovered yet, and it is also possible, due to drip instabilities, that only the lower period 8 elements, up to around element 128, are physically possible.[85][145] No attempts at synthesis have been made for any heavier alkali metals, such as unhexpentium, due to their extremely high atomic number.[36]:1737–1739
Occurrence[edit]
In the Solar System[edit]

Estimated abundances of the chemical elements in the Solar system. Hydrogen and helium are most common, from the Big Bang. The next three elements (lithium, beryllium, and boron) are rare because they are poorly synthesized in the Big Bang and also in stars. The two general trends in the remaining stellar-produced elements are: (1) an alternation of abundance in elements as they have even or odd atomic numbers, and (2) a general decrease in abundance, as elements become heavier. Iron is especially common because it represents the minimum energy nuclide that can be made by fusion of helium in supernovae.[146]
The Oddo–Harkins rule holds that elements with even atomic numbers are more common that those with odd atomic numbers, with the exception of hydrogen. This rule argues that elements with odd atomic numbers have one unpaired proton and are more likely to capture another, thus increasing their atomic number. In elements with even atomic numbers, protons are paired, with each member of the pair offsetting the spin of the other, enhancing stability.[147][148][149] All the alkali metals have odd atomic numbers and they are not as common as the elements with even atomic numbers adjacent to them (the noble gasesand the alkaline earth metals) in the Solar System. The heavier alkali metals are also less abundant than the lighter ones as the alkali metals from rubidium onward can only be synthesized in supernovae and not instellar nucleosynthesis. Lithium is also much less abundant than sodium and potassium as it is poorly synthesized in both Big Bang nucleosynthesis and in stars: the Big Bang could only produce trace quantities of lithium, beryllium and boron due to the absence of a stable nucleus with 5 or 8 nucleons, and stellar nucleosynthesis could only pass this bottleneck by the triple-alpha process, fusing three helium nuclei to form carbon, and skipping over those three elements.[146]
On Earth[edit]

Spodumene, an important lithium mineral
The Earth formed from the same cloud of matter that formed the Sun, but the planets acquired different compositions during the formation and evolution of the solar system. In turn, the natural history of the Earth caused parts of this planet to have differing concentrations of the elements. The mass of the Earth is approximately 5.98×1024 kg. It is composed mostly of iron (32.1%), oxygen (30.1%), silicon (15.1%), magnesium(13.9%), sulfur (2.9%), nickel (1.8%), calcium (1.5%), and aluminium (1.4%); with the remaining 1.2% consisting of trace amounts of other elements. Due to mass segregation, the core region is believed to be primarily composed of iron (88.8%), with smaller amounts of nickel (5.8%), sulfur (4.5%), and less than 1% trace elements.[150]
The alkali metals, due to their high reactivity, do not occur naturally in pure form in nature. They are lithophiles and therefore remain close to the Earth’s surface because they combine readily with oxygen and so associate strongly with silica, forming relatively low-density minerals that do not sink down into the Earth’s core. Potassium, rubidium and caesium are also incompatible elements due to their large ionic radii.[151]
Sodium and potassium are very abundant in earth, both being among the ten most common elements in Earth’s crust;[152][153] sodium makes up approximately 2.6% of the Earth‘s crust measured by weight, making it the sixth most abundant element overall[154] and the most abundant alkali metal. Potassium makes up approximately 1.5% of the Earth’s crust and is the seventh most abundant element.[154] Sodium is found in many different minerals, of which the most common is ordinary salt (sodium chloride), which occurs in vast quantities dissolved in seawater. Other solid deposits include halite, amphibole, cryolite, nitratine, and zeolite.[154] Many of these solid deposits occur as a result of ancient seas evaporating, which still occurs now in places such as Utah‘s Great Salt Lake and the Dead Sea.[6]:69 Despite their near-equal abundance in Earth’s crust, sodium is far more common than potassium in the ocean, both because potassium’s larger size makes its salts less soluble, and because potassium is bound by silicates in soil and what potassium leaches is absorbed far more readily by plant life than sodium.[6]:69
Despite its chemical similarity, lithium typically does not occur together with sodium or potassium due to its smaller size.[6]:69 Due to its relatively low reactivity, it can be found in seawater in large amounts; it is estimated that seawater is approximately 0.14 to 0.25 parts per million (ppm)[155][156] or 25 micromolar.[157] Its diagonal relationship with magnesium often allows it to replace magnesium in ferromagnesium minerals, where its crustal concentration is about 18 ppm, comparable to that of gallium and niobium. Commercially, the most important lithium mineral is spodumene, which occurs in large deposits worldwide.[6]:69
Rubidium is approximately as abundant as zinc and more abundant than copper. It occurs naturally in the minerals leucite, pollucite, carnallite, zinnwaldite, and lepidolite,[158]although none of these contain only rubidium and no other alkali metals.[6]:70 Caesium is more abundant than some commonly known elements, such as antimony, cadmium, tin, and tungsten, but is much less abundant than rubidium.[23]
Francium-223, the only naturally occurring isotope of francium,[9][10] is the product of the alpha decay of actinium-227 and can be found in trace amounts in uranium and thoriumminerals.[159] In a given sample of uranium, there is estimated to be only one francium atom for every 1018 uranium atoms.[160][161] It has been calculated that there is at most 30 g of francium in the earth’s crust at any time, due to its extremely short half-life of 22 minutes.[162][163]
Production and isolation[edit]


Salt flats are rich in lithium, such as these in Salar del Hombre Muerto, Argentina (left) andUyuni, Bolivia (right). The lithium-rich brine is concentrated by pumping it into solar evaporation ponds (visible in Argentina image).
The production of pure alkali metals is difficult due to their extreme reactivity with commonly used substances, such as water. The alkali metals are so reactive that they cannot be displaced by other elements and must be isolated through high-energy methods such as electrolysis.[5][7]
Lithium salts have to be extracted from the water of mineral springs, brine pools, and brine deposits. The metal is produced electrolytically from a mixture of fused lithium chloride and potassium chloride.[164]
Potassium occurs in many minerals, such as sylvite (potassium chloride).[5] It is occasionally produced through separating the potassium from the chlorine in potassium chloride, but is more often produced through electrolysis of potassium hydroxide,[165]found extensively in places such as Canada, Russia, Belarus, Germany, Israel, United States, and Jordan, in a method similar to how sodium was produced in the late 1800s and early 1900s.[166] It can also be produced from seawater. Sodium occurs mostly in seawater and dried seabed,[5] but is now produced through electrolysis of sodium chloride by lowering the melting point of the substance to below 700 °C through the use of a Downs cell.[167][168] Extremely pure sodium can be produced through the thermal decomposition of sodium azide.[169]

For several years in the 1950s and 1960s, a by-product of the potassium production called Alkarb was a main source for rubidium. Alkarb contained 21% rubidium while the rest was potassium and a small fraction of caesium.[170] Today the largest producers of caesium, for example the Tanco Mine, Manitoba, Canada, produce rubidium as by-product from pollucite.[171] Today, a common method for separating rubidium from potassium and caesium is the fractional crystallization of a rubidium and caesium alum (Cs, Rb)Al(SO4)2·12H2O, which yields pure rubidium alum after approximately 30 different reactions.[171][172] The limited applications and the lack of a mineral rich in rubidium limits the production of rubidium compounds to 2 to 4 tonnes per year.[171] Caesium, however, is not produced from the above reaction. Instead, the mining of pollucite ore is the main method of obtaining pure caesium, extracted from the ore mainly by three methods: acid digestion, alkaline decomposition, and direct reduction.[171][173] Both metals are produced as by-products of lithium production: after 1958, when interest in lithium’s thermonuclear properties increased sharply, the production of rubidium and caesium also increased correspondingly.[6]:71
Francium-223, the only naturally occurring isotope of francium,[9][10] is produced naturally as the product of the alpha decay of actinium-227. Francium can be found in trace amounts in uranium and thorium minerals;[159] it has been calculated that at most there are 30 g of francium in the earth’s crust at any given time.[162] As a result of its extreme rarity in nature, most francium is synthesized in the nuclear reaction 197Au + 18O → 210Fr + 5 n, yielding francium-209, francium-210, and francium-211.[174] The greatest quantity of francium ever assembled to date is about 300,000 neutral atoms,[175] which were synthesized using the nuclear reaction given above.[175]
From their silicate ores, all the alkali metals may be obtained the same way: sulfuric acid is first used to dissolve the desired alkali metal ion and aluminium(III) ions from the ore (leaching), whereupon basic precipitation removes aluminium ions from the mixture by precipitating it as the hydroxide. The remaining insoluble alkali metal carbonate is then precipitated selectively; the salt is then dissolved in hydrochloric acid. The result is then left to evaporate and the alkali metal can then be isolated through electrolysis.[7]
Lithium and sodium are typically isolated through electrolysis from their liquid chlorides, with calcium chloride typically added to lower the melting point of the mixture. The heavier alkali metals, however, is more typically isolated in a different way, where a reducing agent (typically sodium for potassium and magnesium or calcium for the heaviest alkali metals) is used to reduce the alkali metal chloride. The liquid or gaseous product (the alkali metal) then undergoes fractional distillation for purification.[7]
Applications[edit]
Lithium, sodium, and potassium have many applications, while rubidium and caesium are very useful in academic contexts but do not have many applications yet.[6]:68 Lithium is often used in batteries, and lithium oxide can help process silica. Lithium can also be used to make lubricating greases, air treatment, and aluminium production.[176]
Pure sodium has many applications, including use in sodium-vapour lamps, which produce very efficient light compared to other types of lighting,[177][178] and can help smooth the surface of other metals.[179][180] Being a strong reducing agent, it is often used to reduce many other metals, such as titanium and zirconium, from their chlorides.[6]:74 Sodium compounds have many applications as well, the most well-known being table salt. Sodium is also used in soap as salts of fatty acids.[181]
Potassium compounds are often used as fertilisers[6]:73[182] as potassium is an important element for plant nutrition. Potassium hydroxide is a very strong base, and is used to control the pH of various substances.[183][184]

FOCS 1, a caesium atomic clock in Switzerland
Rubidium and caesium are often used in atomic clocks.[185] Caesium atomic clocks are extraordinarily accurate; if a clock had been made at the time of the dinosaurs, it would be off by less than four seconds (after 80 million years).[23] For that reason, caesium atoms are used as the definition of the second.[186] Rubidium ions are often used in purple fireworks,[187] and caesium is often used in drilling fluids in the petroleum industry.[23][188]
Francium has no commercial applications,[160][161][189] but because of francium’s relatively simple atomic structure, among other things, it has been used in spectroscopy experiments, leading to more information regarding energy levels and the coupling constants betweensubatomic particles.[190] Studies on the light emitted by laser-trapped francium-210 ions have provided accurate data on transitions between atomic energy levels, similar to those predicted by quantum theory.[191]
Biological role and precautions[edit]

Lithium naturally only occurs in traces in biological systems and has no known biological role, but does have effects on the body when ingested.[192] Lithium carbonate is used as a mood stabiliser in psychiatry to treat bipolar disorder (manic-depression) in daily doses of about 0.5 to 2 grams, although there are side-effects.[192] Excessive ingestion of lithium causes drowsiness, slurred speech and vomiting, among other symptoms,[192] and poisons the central nervous system,[192] which is dangerous as the required dosage of lithium to treat bipolar disorder is only slightly lower than the toxic dosage.[192][193] Its biochemistry, the way it is handled by the human body and studies using rats and goats suggest that it is an essential trace element, although the natural biological function of lithium in humans has yet to be identified.[194][195]
Sodium and potassium occur in all known biological systems, generally functioning as electrolytes inside and outside cells.[196][197] Sodium is an essential nutrient that regulates blood volume, blood pressure, osmotic equilibrium and pH; the minimum physiological requirement for sodium is 500 milligrams per day.[198] Sodium chloride (also known as common salt) is the principal source of sodium in the diet, and is used as seasoning and preservative, such as for pickling and jerky; most of it comes from processed foods.[199] The DRI for sodium is 1.5 grams per day,[200] but most people in the United States consume more than 2.3 grams per day,[201] the minimum amount that promotes hypertension;[202] this in turn causes 7.6 million premature deaths worldwide.[203]
Potassium is the major cation (positive ion) inside animal cells,[196] while sodium is the major cation outside animal cells.[196][197] The concentration differences of these charged particles causes a difference in electric potential between the inside and outside of cells, known as the membrane potential. The balance between potassium and sodium is maintained by ion pumps in the cell membrane.[204] The cell membrane potential created by potassium and sodium ions allows the cell to generate an action potential—a “spike” of electrical discharge. The ability of cells to produce electrical discharge is critical for body functions such as neurotransmission, muscle contraction, and heart function.[204]

A wheel type radiotherapy device which has a long collimator to focus the radiation into a narrow beam. The caesium-137 chloride radioactive source is the blue square, and gamma rays are represented by the beam emerging from the aperture. This was the radiation source involved in the Goiânia accident, containing about 93 grams of caesium-137 chloride.
Rubidium has no known biological role, but may help stimulate metabolism,[205][206][207] and, similarly to caesium,[205][208] replace potassium in the body causing potassium deficiency.[205][207] Caesium compounds are rarely encountered by most people, but most caesium compounds are mildly toxic because of chemical similarity of caesium to potassium, allowing the caesium to replace the potassium in the body, causing potassium deficiency.[208] Exposure to large amounts of caesium compounds can cause hyperirritability andspasms, but as such amounts would not ordinarily be encountered in natural sources, caesium is not a major chemical environmental pollutant.[209] The median lethal dose (LD50) value for caesium chloride in mice is 2.3 g per kilogram, which is comparable to the LD50 values of potassium chloride and sodium chloride.[210]Caesium chloride has been promoted as an alternative cancer therapy,[211] but has been linked to the deaths of over 50 patients, on whom it was used as part of a scientifically unvalidated cancer treatment.[212]Radioisotopes of caesium require special precautions: the improper handling of caesium-137 gamma raysources can lead to release of this radioisotope and radiation injuries. Perhaps the best-known case is the Goiânia accident of 1987, in which an improperly-disposed-of radiation therapy system from an abandoned clinic in the city of Goiânia, Brazil, was scavenged from a junkyard, and the glowing caesium salt sold to curious, uneducated buyers. This led to four deaths and serious injuries from radiation exposure. Together with caesium-134, iodine-131, and strontium-90, caesium-137 was among the isotopes distributed by the Chernobyl disaster which constitute the greatest risk to health.[48]
Francium has no biological role[213] and is most likely to be toxic due to its extreme radioactivity, causing radiation poisoning,[214] but since the greatest quantity of francium ever assembled to date is about 300,000 neutral atoms,[175] it is unlikely that most people will ever encounter francium.
Radium Girls
From Wikipedia, the free encyclopedia
This article is about the factory workers. For the illusion, see Radium Girl.

Radium dial painters working in a factory
The Radium Girls were female factory workers who contracted radiation poisoning from painting watch dials withself-luminous paint at the United States Radium factory in Orange, New Jersey, around 1917. The women, who had been told the paint was harmless, ingested deadly amounts of radium by licking their paintbrushes to give them a fine point; some also painted their fingernails and teeth with the glowing substance.
Five of the women challenged their employer in a case that established the right of individual workers who contractoccupational diseases to sue their employers.
United States Radium Corporation[edit]
Main article: United States Radium Corporation
_advertisement,_1921,_retouched.png)
1921 advertisement for Undark
From 1917 to 1926, U.S. Radium Corporation, originally called the Radium Luminous Material Corporation, was engaged in the extraction and purification of radium from carnotite ore to produce luminous paints, which were marketed under the brand name “Undark“. As a defense contractor, U.S. Radium was a major supplier of radioluminescent watches to the military. Their plant in Ottawa, Illinois, employed more than a hundred workers, mainly women, to paint radium-lit watch faces and instruments, misleading them that it was safe.
Radiation exposure[edit]
The U.S. Radium Corporation hired approximately 70 women to perform various tasks including the handling of radium, while the owners and the scientists familiar with the effects of radium carefully avoided any exposure to it themselves; chemists at the plant used lead screens, masks and tongs.[1] U.S. Radium had even distributed literature to the medical community describing the “injurious effects” of radium.
An estimated 4,000 workers were hired by corporations in the U.S. and Canada to paint watch faces with radium. They mixed glue, water and radium powder, and then used camel hair brushes to apply the glowing paint onto dials. The then-current rate of pay, for painting 250 dials a day, was about a penny and a half per dial (equivalent to $0.276 in 2015). The brushes would lose shape after a few strokes, so the U.S. Radium supervisors encouraged their workers to point the brushes with their lips, or use their tongues to keep them sharp. For fun, the Radium Girls painted their nails, teeth and faces with the deadly paint produced at the factory.[2] Many of the workers became sick. It is unknown how many died from exposure to radiation.
Radiation sickness[edit]
Many of the women later began to suffer from anemia, bone fractures and necrosis of the jaw, a condition now known as radium jaw. It is thought that the X-ray machines used by the medical investigators may have contributed to some of the sickened workers’ ill-health by subjecting them to additional radiation. It turned out at least one of the examinations was a ruse, part of a campaign of disinformation started by the defense contractor.[1] U.S. Radium and other watch-dial companies rejected claims that the afflicted workers were suffering from exposure to radium. For some time, doctors, dentists, and researchers complied with requests from the companies not to release their data. At the urging of the companies, worker deaths were attributed by medical professionals to other causes; syphilis, a notorious sexually transmitted disease at the time, was often cited in attempts to smear the reputations of the women.[3]
Significance[edit]
Litigation[edit]
The story of the abuse perpetrated against the workers is distinguished from most such cases by the fact that the ensuing litigation was covered widely by the media. Plant worker Grace Fryer decided to sue, but it took two years for her to find a lawyer willing to take on U.S. Radium. Even after the women found a lawyer, the slow-moving courts held out for months. At their first appearance in court on January 1928, two women were bedridden and none of them could raise their arms to take oath. A total of five factory workers – Grace Fryer, Edna Hussman, Katherine Schaub, and sisters Quinta McDonald and Albina Larice – dubbed the Radium Girls, joined the suit. The litigation and media sensation surrounding the case established legal precedents and triggered the enactment of regulations governing labor safety standards, including a baseline of “provable suffering“.
Historical impact[edit]
The Radium Girls saga holds an important place in the history of both the field of health physics and the labor rights movement.[citation needed] The right of individual workers to sue for damages from corporations due to labor abuse was established as a result of the Radium Girls case.[citation needed] In the wake of the case, industrial safety standards were demonstrably enhanced for many decades.[citation needed]
The case was settled in the autumn of 1928, before the trial was deliberated by the jury, and the settlement for each of the Radium Girls was $10,000 (equivalent to $137,000 in 2015) and a $600 per year annuity (equivalent to $8,200 in 2015) while they lived, and all medical and legal expenses incurred would also be paid by the company.[4][5]
The lawsuit and resulting publicity was a factor in the establishment of occupational disease labor law.[6] Radium dial painters were instructed in proper safety precautions and provided with protective gear; in particular, they no longer shaped paint brushes by lip and avoided ingesting or breathing the paint. Radium paint was still used in dials as late as the 1960s.[7]

Former factory site
Scientific impact[edit]
Robley D. Evans made the first measurements of exhaled radon and radium excretion from a former dial painter in 1933. At MIT he gathered dependable body content measurements from 27 dial painters. This information was used in 1941 by the National Bureau of Standards to establish the tolerance level for radium of 0.1 μCi (3.7 kBq).
The Center for Human Radiobiology was established at Argonne National Laboratory in 1968. The primary purpose of the Center was providing medical examinations for living dial painters. The project also focused on collection of information and, in some cases, tissue samples from the radium dial painters. When the project ended in 1993, detailed information of 2,403 cases had been collected. No symptoms were observed in those dial painter cases with less than 1,000 times the natural 226Ra levels found in unexposed individuals, suggesting a threshold for radium-induced malignancies.[8]
Literature and film[edit]
- The story is told in Eleanor Swanson’s poem Radium Girls, collected in A Thousand Bonds: Marie Curie and the Discovery of Radium (2003, ISBN 0-9671810-7-0)
- D. W. Gregory told the story of Grace Fryer in the play Radium Girls, which premiered in 2000 at the Playwrights Theatre in Madison, New Jersey.
- There is an elaborate reference to the story in the Kurt Vonnegut novel Jailbird (1979, ISBN 0-385-33390-0)
- Poet Lavinia Greenlaw has written on the subject in The Innocence of Radium (Night Photograph, 1994)
- Historian Claudia Clark wrote an account of the case and its wider historical implications: Radium Girls: Women and Industrial Health Reform, 1910–1935 (published 1997).
- Ross Mullner’s book Deadly Glow: The Radium Dial Worker Tragedy describes many of the events (1999, ISBN 0-87553-245-4)
- The story is told by Jo Lawrence in her short animated film “Glow” (2007)
- The story is referenced in the 2006 film Pu-239
- The Michael A. Martone short story It’s Time is told from the perspective of an unnamed Radium Girl
- A fictionalized version of the story was featured in the Spike TV show 1000 Ways to Die (#196)[9] and Science Channel‘s Dark Matters: Twisted But True
- Radium Halos: A Novel about the Radium Dial Painters a 2009 novel by Shelley Stout is historical fiction narrated by a sixty-five-year-old mental patient who worked at the factory when she was sixteen (ISBN 978-1448696222).
- Author Deborah Blum referenced the story in her 2010 book The Poisoner’s Handbook: Murder and the Birth of Forensic Medicine in Jazz Age New York.
- Author Robert R. Johnson features a story on the radium girls in his book Romancing the Atom. (ISBN 978-0313392795) [10]
- The Case of the Living Dead Women, a website displaying scans of 180 pages of newspaper clippings about a similar incident, the Ottawa, Illinois Radium Dial Companylitigation[11]
- The Radium Dial Company workers’ story is dramatized in Melanie Marnich‘s stage play These Shining Lives.
- A fictionalized version of the story was featured in the 1937 short story “Letter to the Editor” by James H. Street, adapted into a 1937 film Nothing Sacred and a 1953 Broadway musical Hazel Flagg.
- The story is told in the American Experience episode The Poisoner’s Handbook, based on the non-fiction book with the same name by Deborah Blum.
Mystery Balls Found In Atlantic, Small Metal Orbs Found In Earth’s Atmosphere
Does metal found in a 2,600-year-old shipwreck prove that Atlantis DID exist? Mythical red alloy said to be from the lost island is discovered off coast of Sicily
A mythical metal said by ancient Greeks to be found in the lost city of Atlantis has been recovered from a ship that sunk 2,600 years ago off the coast of Sicily.
Marine archaeologists found 39 ingots of what they believe is ‘orichalcum’ on the sandy seabed among the wreck of a trading vessel that sank 1,000 feet off the coast of the town of Gela, in sourthern Sicily.
The wreck is the fifth ancient ship to be recovered off the coast of the town.
Scroll down for video

This map of Atlantis – oriented with south at the top – was drawn by 17th century scholar Athanasius Kircher, who pinpointed the mythical continent as being in the mid-Atlantic before it was lost to the sea
It is thought that it had been carrying cargo from either Greece or Asia Minor when it was caught in a storm and sunk.
Professor Sebastiano Tusa, an archaeologist at the office of the Superintendent of the Sea in Sicily, claimed the metal they had discovered in the remains of the ship was probably the mythical and highly prized red metal orichalcum.
Analysis of the metal ingots revealed they were made from an alloy of copper and zinc with traces of nickel, lead and iron.
Speaking to Discovery News, Professor Tusa said: ‘Nothing similar has ever been found.
‘We knew orichalcum from ancient texts and a few ornamental objects.
‘The wreck dates to the first half of the sixth century.
‘It was found about 1,000 feet from Gela’s coast at a depth of 10 feet.’
If the metal discovered by Professor Tusa and his team is really the mythical orichalcum, then it lends support to the idea of Atlantis as being a real place.
The existence of the island is greatly debated among historians and archaeologists.
Some believe it is entirely fictional while others claim stories of the ‘Island of Atlas’ may have been based on a real historical location that was drowned by rising sea levels or a tsunami.
The Egyptian city of Heracleion, for example, was lost 1,200 years ago when it was engulfed by the sea.
Most of the legend of Atlantis comes from the work of the Greek philosopher Plato, who describes how the great nation was submerged beneath the Atlantic Ocean after falling out of favour with the Gods.
Plato mentions orichalcum in the Critias dialogue and describes Atlantis as flashing with the ‘red light’ of the metal.
He wrote that orichalcum was highly prized and second only in value to gold. It was mined in the mythical island and covered the surfaces of Poseidon’s temple.
The existence of this metal and its composition has since been widely debated, but it is commonly thought to be a brass-like alloy. Brass is made from copper and zinc.
It is thought to have been made through a process called cementation, which reacts zinc ore with charcoal and copper in a crucible.
X-ray fluorescence of the ingots found off the coast of Gela show they were made from 75-80 per cent copper, 15-20 per cent zinc and small amounts of nickel, lead and iron.
Professor Tusa said: ‘The finding confirms that about a century after its foundation in 689BC, Gela grew to become a wealthy city with artisan workshops specialised in the production of prized artifacts.’

Professor Sebastiano Tusa, centre, and his team of divers discovered the metal ignots of what they believe to be the mythical metal of orichalcum on a 2,600-year-old shipwreck found off the coast of Gela in Sicily

The shipwreck with the ingots was found 1,000 feet off shore of the town of Gela in the sourthern part of Sicily

Statues like this one above from the sunken Egyptian city of Heracleion have recently been rediscovered by marine archaeologists, raising hopes that if Atlantis did ever exist then it may still be found under the sea

However, some experts disagree about the composition of orichalcum. Enrico Mattievich, a former physics professor at the Federal University of Rio de Janeiro, believes the metal has its origins in the Chavin civilisation that developed in the Peruvian Andes in around 1,200 BC.
He claims the metal alloy is made from copper, gold and silver.
He claims that the discovery off the coast of Gela is not true orichalcum.
Indeed in Ancient Rome, coins made from brass were also said to be made from orichalcum.

The lost civilisation of Atlantis was reputed to have many treasures including buildings clad in the metal orichalcum that were lost to the sea after the people there apparently fell out of favour with the gods

There have been many locations around the world suggested as potential sites for the lost island of Atlantis
